Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jingzhi Fan | + 5099 word(s) | 5099 | 2021-07-05 05:52:25 | | | |
| 2 | Bruce Ren | -21 word(s) | 5078 | 2021-07-12 10:22:37 | | | | |
| 3 | Dean Liu | Meta information modification | 5078 | 2021-09-28 06:09:59 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Fan, J. Metabolomics in Bone Research. Encyclopedia. Available online: https://encyclopedia.pub/entry/11955 (accessed on 24 June 2026).
Fan J. Metabolomics in Bone Research. Encyclopedia. Available at: https://encyclopedia.pub/entry/11955. Accessed June 24, 2026.
Fan, Jingzhi. "Metabolomics in Bone Research" Encyclopedia, https://encyclopedia.pub/entry/11955 (accessed June 24, 2026).
Fan, J. (2021, July 12). Metabolomics in Bone Research. In Encyclopedia. https://encyclopedia.pub/entry/11955
Fan, Jingzhi. "Metabolomics in Bone Research." Encyclopedia. Web. 12 July, 2021.
Copy Citation
Identifying the changes in endogenous metabolites in response to intrinsic and extrinsic factors has excellent potential to obtain an understanding of cells, biofluids, tissues, or organisms’ functions and interactions with the environment. The advantages provided by the metabolomics strategy have promoted studies in bone research fields, including an understanding of bone cell behaviors, diagnosis and prognosis of diseases, and the development of treatment methods such as implanted biomaterials.
metabolomics
bone homeostasis
osteoporosis
bone regeneration
osteosarcoma
biomaterials
1. Introduction
Bone is the primary tissue in the skeleton system. At first glance, bone tissue might appear to be a static and rugged part of the body with almost no dynamics. However, the first impression is deceiving. Bone homeostasis is a very dynamic and complex process closely linked to other organism functions, including immunity. Moreover, the disturbances in these processes are associated with numerous diseases, chronic or acute, that severely affect life quality and are even life-threatening.
Among them, osteoporosis, the most well-known bone disorder, has a high prevalence rate, and the rate for osteopenia is even higher [1]. This chronic bone disease has a preference among postmenopausal women (over 10%) and the aged population [2][3], and it highly increases the risk of osteoporotic fracture. Pathologic fracture can also arise from weakened bone caused by tumors. Bone fracture, including traumatic and pathological caused, is a malignant medical condition that frequently happens for various reasons. The loss of productivity and individual disability after fracture dictates a high demand for hard tissue healing studies. It has become evident that a much deeper understanding of hard tissue and related diseases is needed to develop a novel diagnosis, prognosis, and treatment approaches. The need for this will only increase due to the ageing population.
In this regard, metabolomics—a large-scale study of metabolites that are directly involved in biochemical activity holds great promise to advance our knowledge on bone tissue (patho)physiology. Describing the metabolite pathways involved in a disease can greatly promote understanding disease development, diagnosis, and prognosis [4]. Moreover, the prominent biomarker identification can be incorporated with the traditional diagnostic methods to improve diagnostic accuracy. Additionally, metabolite ability to modulate phenotype directly through biochemical reaction can pave the road for developing new treatment approaches, especially blockers, competitive inhibitors, and accelerants.
In recent years, there has been a growing interest in metabolomics for bone tissue-related research, which has resulted in an improved understanding of underlying cellular processes during bone homeostasis and disease. However, the available information about bone–immune system and cell–cell interactions is still sparse and systematic studies are needed to deepen our understanding about these interactions. This review provides an overview of the metabolic processes relevant to bone homeostasis and the most prevalent bone diseases. It summarizes recent studies that employ metabolomics to better understand bone metabolism (Figure 1B). With this review, we want to showcase the role and value of metabolomics in bone research and encourage incorporating metabolomics in more bone-related studies, as it is providing unique insights.
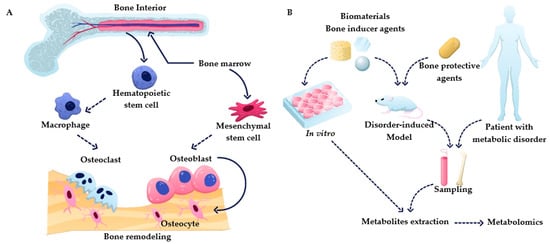
Figure 1. (A) Overview of bone remodeling processes, including osteogenesis and osteoclastogenesis. (B) Summary of metabolomics applications in bone research.
2. Metabolism of Bone Cells
2.1. Bone Homeostasis
Bone is the primary tissue in the skeleton system, providing the function of structure support, hematopoiesis location, and mineral storage. To perform these functions, bone has a complex structure that contains compact bone and cancellous bone, with the composition of organic and inorganic composites containing living cells [5]. The bone remodeling process is a cascade of regular activities in which three cell types, including osteoblasts, osteoclasts, and osteocytes, play crucial roles (shown in Figure 1). Bone undergoes continuous resorption carried out by osteoclasts and formation by osteoblasts, which are linked cell activities that form a dynamic equilibrium in bone tissue [6]. To briefly summarize this complex process, osteoclast precursors are activated to start resorption stages by cytokines such as receptor activator of nuclear factor-kB (RANK) and macrophage colony-stimulating factor (M-CSF) released by bone lining cells upon receiving a biochemical signal such as hormones (e.g., parathyroid) or mechanical signal (due to bone damage). Osteoclasts, specialized cells developed from the monocyte/macrophage lineage, can contact the bone matrix to form a sealed bone resorption area, playing a role in degrading bone tissue [7]. The activity of the osteoclast is terminated by the formation of resorption pits on the bone surface. During the formation process, osteoblasts deposit the osteoid into the resorbed site to form a mineralized matrix. Osteoblasts are then either apoptotic or buried as osteocytes in new bone layers. It is crucial to maintain a balance between osteoclasts and osteoblasts. Bone remodeling is a dynamic and lifelong process. An imbalance in this process leads to bone disorders. It can cause several diseases, such as overactive osteoclasts and bone turnover, which would lead to osteopenia and osteoporosis, while abnormal osteoblasts elevate the risks for hyperosteogeny and tumor [3][8].
Cell differentiation is an integral part of bone homeostasis, as the osteoclast is derived from hematopoietic cells, while osteoblasts are produced from the differentiation of bone marrow stem cells (MSCs). The differentiation from MSC to osteoblast and from monocyte-macrophage lineage to osteoclast involves metabolic changes and adaptions. Metabolomics is one of the key methodologies to study these processes and deepen the understanding of osteogenesis and osteoclastogenesis and their relationship with bone diseases. MSCs, the precursor of osteoblasts, predominantly utilize glycolysis as the energy source within the bone marrow; however, the energy metabolism shifts to oxidative phosphorylation during the differentiation [9][10]. Hypoxia-inducible factor 1 α (HIF-1α) works as a regulator of MSC energy metabolism during osteogenic and chondrogenic differentiation [11]. HIF-1α can promote glycolysis and inhibit oxidative phosphorylation (OxPhos) and is most active in undifferentiated MSCs [12]. MSCs have been shown to grow and proliferate in the glycolysis pathway in hypoxic condition. At the same time, they undergo a metabolic shift to the TCA pathway to supply the required energy to differentiate into the bone cells [9]. As a result, a higher OxPhos activity with an unaltered or decrease in glycolysis happens in MSCs differentiation [9][12][13]. In mature osteoblasts, glycolysis produces most of the needed energy for osteoblasts to maintain the functionality [14]. The well-known osteogenic factor, bone morphogenetic protein, can boost glucose metabolism and therefore enhance the development of bone and cartilage [15].
During osteoclastogenesis, both glycolysis and OxPhos were found increased, as indicated with a higher glucose consumption and lactate production [16]. This means that glycolysis and mitochondrial processing both play a role in the differentiation and absorption. Increased expression of glycolytic genes, including hexokinase, phosphofructokinase, and pyruvate kinase, was found during osteoclastogenesis [17]. The final osteoclasts may become apoptosed, which makes them difficult to study [18]. However, there is some evidence which indicates an alternative fate for RANKL-stimulated osteoclasts-osteomorphs production, [19]. The lifespan of osteoclasts is generally considered to be as long as 2 weeks [20]. Nevertheless, Jacome-Galarza et al. described a mechanism upon which osteoclasts could be maintained in bone, leading to compensate for osteoclast deficiency in vivo [21].
Lipid metabolism can also alter the differentiation and function of bone cells. Long-chain polyunsaturated fatty acids, such as docosahexaenoic acid and arachidonic acid, can directly suppress the differentiation and activity of osteoclasts [22]. As part of lipid rafts to control the signal transduction during osteoclastogenesis, cholesterol can alter the RANK-RANKL signal transduction, resulting in increased bone resorption under a high content of cholesterol [23]. Similarly, phosphoinositide, a membrane lipid, regulates calcium signaling and influences osteoclast differentiation [23]. The changes of 18 metabolites, most of which were lysophosphatidylcholines, were detected in the study of differentiation inhibition with estradiol [24]. The enzymatic expression changes explained this inhibition during osteoclast differentiation and oxidative/anti-oxidative imbalance during osteoclast proliferation. It also should be pointed out that lipids are used as an energy source which in turn can influence cell functions. Low lipid levels in the skeletal stem cells environment leads to chondrogenesis rather than osteogenesis due to the osteoblasts dependency on the fatty acid oxidation (FAO) [25].
The metabolism studies on osteogenesis and osteoclastogenesis provide a guide to regulate the differentiation of precursor cells by altering the metabolic pathways. Monitoring the cell function upon exposure to a change in its surrounding environment allows for controlling cells’ behavior. Osteogenesis differentiation from MSCs can be guided by parathyroid hormone by enhancing glycolysis to increase bone formation [24]. Lee et al. attempted to regulate adipocyte and osteoblast differentiation from MSCs using several metabolites. Among them, ergosterol peroxide and 9,11-dehydroergosterol peroxide were found to inhibit the differentiation toward adipocytes. Diterpenes dehydroabietic acid, 7-oxocallitrisic acid, and pimaric acid, on the other hand, induced osteoblasts differentiation [24]. The effect of high and low amounts of energy source, oxygen, and ROS as environmental factors on a deviation of cellular behavior has been well reviewed recently [6].
2.2. Osteoimmunology
The close connection between the immune system and bone tissue generated the conception of osteoimmunology [26]. Immune cells have a remarkable effect on bone physiology by acting on osteogenesis and osteoclastogenesis. The activity of bone resorption is also regulated by interleukins, cytokines secreted by immune cells. Among all immune cells, macrophages play an essential regulatory role in bone regeneration. Macrophages are divided into two polarized extremes, pro-inflammatory M1 and anti-inflammatory M2 [27]. Extracellular amino acid levels can alter the immune system functions through altering macrophage phenotypes. Arginine (Arg), ornithine, tryptophan, and glutamine can be considered amino acid-based immunomodulators [28]. Arginine metabolism plays a critical role in macrophage polarization and characterization. The prominent metabolic feature of M1 macrophages is Arg catabolism to nitric oxide (NO) and citrulline using nitric oxide synthase (NOS). While for M2 macrophages, Arg is catabolized to ornithine and urea through arginase [27]. Therefore, the catabolic and anabolic activity of NOS and arginase processes can regulate the polarization of macrophage and thus influence the inflammation and regeneration in hard tissue [29]. Indeed, the chronic inflammation of the target tissue can be pharmacologically controlled by regulating NO and Arg production [30].
In bone regeneration processes, activation of M1 macrophages in the early inflammatory phase leads to the secretion of inflammatory cytokines such as tumor necrosis factor α (TNF-α), Interleukin 6 (IL-6), and IL-1b, which results in activation of the osteoclastogenesis cascade with consequential bone resorption [31]. The M1 macrophage effect on mesenchymal stem cell (MSC) osteogenesis through the COX-2-prostaglandin E2 pathway has also been reported [32]. M2 macrophages are involved in the secondary stages of bone repair, which can lead to fibrous capsule or bone formation. Fibrous capsule formation prevents bone marrow stem cells being connected to the surface of a biomaterial for further progress in osteogenesis. Therefore, the domination of the inflammatory phase of macrophages guarantees the long-term failure or success of bone regeneration in the subsequent stages. The protracted stage of the M1 phenotype results in more fibrous inducible cytokines are amplified and fibrous capsules are formed, helping M2 phenotypes [31]. The immune response elicited by primary M1 macrophages in the early stages determines the decision of secondary M2 macrophages to secrete specific cytokines. In summary, macrophages positively contribute to bone healing by secretion of osteogenesis-related growth factors or negatively interfering through fibrous-induced inflammatory cytokines.
From an energy metabolism point of view, macrophages are constantly switching from glycolysis to oxidative phosphorylation (OxPhos). It was proved that M1 phenotypes use glycolysis and pentose phosphate pathways to uptake required ATP. M1 phenotypes lead to metabolic perturbations in several metabolites of the TCA cycle, including citrate, succinate, and itaconate. Prolonged M1 polarization can be selected as a metabolic marker for the diagnosis of inflammatory diseases such as rheumatoid arthritis and metabolic disorders such as diabetes and osteoporosis [28]. On the contrary, glycolysis has been shown to not be relevant for M2 polarization, while the consumption of glutamine Gln can be upregulated to fuel the TCA cycle [33]. Elevated FAO and OxPhos pathways indicate M2 macrophage activity and sustain the inflammatory response [27]. Gln metabolism has been reported as the synergistic support for macrophage activation through M2 phenotype differentiation [34]. Macrophages have the potential to uptake different types of lipids, including VLD, LDL, and oxidized lipoproteins. Lysosomes convert the consumed lipids to free fatty acids and cholesterol, which eventually are involved in OxPhos and ETC pathways. As lipid-based macrophage modulators, fatty acid synthesis (FAS) and FAO guide M1 and M2 macrophage polarization, respectively [28]. This suggests that the tracking of metabolites involved in macrophage energy homeostasis can serve as a marker to evaluate a transition from the M1 phenotype towards M2. In fact, it has been demonstrated that metabolomics, through an elucidation of changes in macrophage polarization, provides accurate information about an inflammatory disease [28][35].
3. Metabolomics in Research of Bone Diseases
As demonstrated in the previous section, metabolism plays a crucial role in bone homeostasis, and disturbances can cause or are caused by several diseases. Therefore, it is evident that metabolite measurements could provide a valuable readout for bone disease research. Indeed, metabolomics has been used for the identification of possible biomarkers for the diagnosis and prediction of various bone-related diseases. Besides diagnosis, metabolomics has also been applied to monitor the treatment. Perturbations in several major metabolic pathways due to bone diseases have been reported. Specifically, arginine and its related metabolism pathways are critical in osteoimmunology. Energy metabolisms, including glycolysis and TCA cycle, are pivotal for bone cell differentiation and function. PUFAs are valuable for dietetic, clinical, and biological research of osteoporosis and arthritis. Furthermore, fatty acids, such as arachidonic acid, are exploitable to develop advanced treatments. The metabolites related to acute and chronic inflammation are also closely involved in bone defects. Additionally, energy-related metabolites such as lactate and glutamine are altered in bone cancer. These metabolites and pathways can be utilized to develop diagnosis, prognosis, and treatment procedures of bone diseases.
3.1. Osteoporosis
Osteoporosis (OP) is the most known and common bone disease. OP occurs worldwide in all populations, with a higher prevalence among postmenopausal women and aged people. A low bone mineral density, which increases fracture risk, is the hallmark of OP. OP is usually characterized by metabolic disorders of bone tissue, and usage of metabolomics to study pathophysiology has gained popularity [36]. Animal models are commonly used to study the molecular mechanisms of OP in vivo. Ovariectomized (OVX) mice is an animal model of OP that mimics postmenopausal women with low BMD. Using this model, ovarian extraction results in a decrease in estrogen and progesterone, with consequences elevating the rate of bone loss [36]. Lipid metabolisms, especially arachidonic acid metabolism, linoleic acid metabolism, and glycerophospholipid metabolism, are impacted by the decrease in estrogen [37][38]. Polyunsaturated fatty acids (PUFAs) are well known for their influence on BMD [39]. Two different families of PUFAs, n-3 PUFA, derived from α- linolenic acid, and n-6 PUFA, derived from linoleic acid, are frequently occurring in metabolomics studies with their opponent functions [22]. In fact, elevated levels of arachidonic acid (AA), an n-6 PUFA, have been observed in oophorectomized rats and postmenopausal women with OP [37][38][40]. Arachidonic acid can stimulate the expression of receptor activator of NF-kB ligand (RANKL), leading to a high plasma RANKL level [22]. As the cytokine essential for osteoclast differentiation, a high concentration of RANKL can remarkably promote osteoclastogenesis, resulting in the loss of bone tissue [41]. On the contrary, docosahexaenoic acid (DHA), an n-3 PUFA, suppresses osteoclast formation from human CD14-positive monocytes by the reduction of key signaling transduction pathways of kinases (JNK, ERK, and p38 MAPKs) [22][42]. Thus, DHA inhibits osteoclastogenesis by blocking RANKL-induced activation from primary macrophages. At the same time, prostaglandin and leukotrienes, the downstream metabolic products of arachidonic acid, were also proved to impact Wnt signaling (osteoblastogenesis) and osteoclastic resorption, respectively [43][44]. A high-level accumulation of other lipid metabolites has been found in postmenopausal women and oophorectomized rats as well [45][46][47]. Lipid metabolisms not only impact osteoclastogenesis but also influence osteoblastogenesis. The increased lipid oxidation, followed by high oxidative stress, can activate peroxisome proliferator-activated receptor γ, consequently inhibiting osteoblastogenesis and promoting adipogenesis [48]. PUFAs also play various roles in osteoblastogenesis. Fatty acids can activate peroxisome proliferator-activated receptor γ (PPARγ) [49]. PPARγ plays a pivotal part in cell-fate determination, guiding the MSCs to differentiate into adipocytes [50]. This could explain the bone loss of the patients with a high-fat diet. AA decreases the expression of osteogenic markers and the osteoprotegerin/RANKL ratio, causing the appearance of adipocytes in MSC differentiating during osteoblastogenesis [51]. However, n-3 PUFA such as DHA and eicosapentaenoic acid do not have such an impact on MSCs [51][52]. In an attempt to characterize the pathological mechanism of the postmenopausal OP, metabolomics was employed to analyze the OVX mice-related femur tissue. The obtained data indicated altered levels of 93 lipid metabolites such as fatty acyls, glycerolipids, glycerophospholipids, sphingolipids, and sterols, among which levels of many fatty acids were increased in the OVX model [53]. To sum up, PUFAs play a critical role in osteoporosis by (1) promoting osteoclastogenesis through the expression of RANKL and (2) altering the differentiation of MSCs by inducing adipogenesis.
Ageing is another leading cause of low BMD. The fractures due to senile OP are highly life-threatening for the aged population, especially those over the age of 70 [1]. It is known that the functionality of osteoblasts, adipocytes, and osteoclasts changes with ageing. A higher adipogenesis level was estimated with the accumulation of bone marrow fat, not found in postmenopausal OP, which may be a critical cause for lower osteoclastogenic activity [54][55]. An in vitro study verified that adipocytes, with their metabolites, inhibit osteoblast differentiation by downregulating histone acetylation [56]. Again, lipid metabolism appears to have an essential role in the age-associated reduction of BMD, as demonstrated by a metabolomics study on senescent osteoblasts [57]. The authors identified the downregulation of n-3 PUFAs in the fatty acid metabolism due to the severe oxidative stress damage. The decrease in n-3 PUFAs, as explained before, could result in an elevation of bone loss through osteoclasts.
At the clinical level, a plasma metabolites profiling conducted on 1552 individuals to identify BMD-associated metabolomic markers detected a higher level of creatine, dimethylglycine, and glycine [58]. The authors suggested that these metabolites were causally negatively associated with BMD by altering bioenergetic processes as well as glycine and threonine metabolism pathways. As a major component of the protein collagen, hydroxyproline is also considered an OP-specific marker [59]. Serum metabolite components of postmenopausal women with low BMD were investigated using CE-MS and showed that the level of hydroxyproline could be a marker of OP [60]. Increased concentration of hydroxyproline indicates the degradation of collagen type I from the bone matrix and it was reduced after treatments [61]. Similarly, other catabolites developed in bone from collagen, such as deoxypyridinoline (collagen stabilizer) and pyridinoline (cross-linking compound of collagen fibers), can also serve as the markers of OP [59]. However, changes in their levels are the results rather than the causes of OP; therefore, they are more suitable for the development of diagnosis but not treatment.
3.2. Bone Injuries
Bone fractures have always been a relevant threat to public health worldwide. Several studies have recently been published investigating the metabolic processes during both inflammatory response and tissue regeneration after bone fractures.
Ibrahim et al. identified the changes in the plasma metabolite levels in patients who went through intramedullary reaming [62]. The precursors to extracellular matrix proteins, lipids, and cysteine displayed remarkable elevation after the surgery. At the same time, the abundance of tryptophan decreased. Galactosamine and glucuronic acid, as the precursors of chondroitin sulphate, were elevated in the after-reaming plasma. The precursors of hyaluronic acid, such as acetylglucosamine, were also increased. The increase in these ECM building blocks indicates that the matrix remodeling is accelerated after the surgery. The increased lipids, including ceramides, myristoleate, and phosphoethanolamine, play well-recognized roles in inflammation. They modify inflammatory processes by impacting inflammatory cell signaling and gene expression patterns [63]. Additionally, glutathione and taurine, the downstream products of cysteine, are both antioxidants, protecting the cells from reactive oxygen species [64][65]. At the same time, 4-hydroxynonenal, which contributes to oxidative stress, was reduced after the reaming [66]. Linolenate, a precursor of arachidonate (an inflammatory mediator), was found to be increased, much like corticosterone and cortisol, which are inhibitors in the proinflammatory pathway [67]. Kynurenine, a metabolite of tryptophan, was elevated in the post-reaming plasma, while tryptophan itself declined, pointing towards an upregulated activity of the kynurenine pathway [68][69]. It should be pointed out that the increase in the kynurenine/tryptophan ratio has been considered a marker for indoleamine 2,3-dioxygenase (IDO) activity [69]. Thus, based on the anti-inflammatory mechanism of IDO, the increase in kynurenine benefits anti-inflammatory processes as well [70]. Due to the anti-inflammatory function, IDO is considered a critical regulator in graft-versus-host disease, and as such the mediation of IDO is important for implantable biomaterials [71]. Additionally, kynurenic acid, a metabolite of kynurenine through transferase, decreased the release of TNF-α, further reducing inflammation response [69][72]. Taking it all together, the metabolites profiling after surgery indicated that the body tends to reduce the inflammation response after wound healing.
Bone turnover markers, including the biomarkers of bone resorption, bone formation, and osteoclast regulatory proteins, have been utilized to evaluate bone homeostasis. As a result, the measurement of these markers can be employed to monitor the healing of fractured bones [73]. Veitch et al. detected the alterations of these markers following tibial shaft fracture in 24 weeks [74]. Both bone resorption and formation markers were elevated, which was in line with the observation from the animal model study using sheep conducted by Sousa et al. [75]. In the first two weeks, the catabolites of type-I collagen-like C-terminal telopeptide (bone resorption marker) had a higher increase compared to the bone formation markers, such as bone alkaline phosphatase and osteocalcin [74]. This highlighted that the activity of necrotic tissue resorption is high at the beginning stage of wound healing, following the reconstruction of bone tissue.
The metabolic signatures provide valuable clues to establish “the big picture” on bone regeneration from critical injury. There are seemingly microscopic but significant differences in defect and injury types; however, only a few studies are available. Therefore, further metabolite profiling studies considering all aspects of bone injuries are needed to understand the molecular mechanisms affected by injury and healing.
3.3. Rheumatoid Arthritis
Rheumatoid arthritis (RA) is commonly considered arthritis driven by autoimmune pathogenesis. The analysis of joint tissue metabolism, especially with immune cells, can ameliorate the understanding of pathogenesis and prognosis. The bone destruction of rheumatoid arthritis happens mainly due to the abnormal activation of osteoclasts [76]. Therefore, the overactivity of osteoclasts stimulated by various factors may cause and/or aggravate arthritis. Similar to the PUFAs in osteoporosis, n-6 PUFAs, as the precursor of inflammatory eicosanoids, are responsible for the cartilage loss and inflammation in RA [77]. The influence of n-3/n-6 PUFA intake on RA has already been extensively studied among dietary studies and clinical medicine, which suggest increasing the intake of n-3 PUFAs such as DHA and to avoid the intake of n-6 PUFAs such as AA [63][78][79]. Another fatty acid family, short-chain fatty acid (SCFA), is also relevant for arthritis. Lower butyrate (SCFA) levels in the blood of RA patients and arthritic mice have been reported [80]. The amelioration of the severity of systemic autoimmune inflammatory conditions was achieved by oral administration of SCFAs [81]. Furthermore, the butyrate ability to suppress the expression of inflammatory cytokines from T cells by promoting the expression of IL-10 has been demonstrated [82].
Energy metabolism is also abnormal in RA. Narasimhan et al. performed NMR-based metabolite profiling of the serum from patients with rheumatoid arthritis to identify the synovial joint biomarkers [83]. The metabolites with abnormal levels found in RA serum were associated with the TCA cycles, fatty acid, and amino acid metabolism. Among them, lactate and pyruvate were significantly upregulated. This can be explained by the higher bioenergetic and biosynthetic demands in the inflamed tissue [83]. The RA metabolic process of T cells that drives tissue inflammation could be attributed to the mitochondrial defect [84]. The mitochondrial DNA containments trigger T cell tissue invasion in the organelle assembled inflammasome and caspase-1. Another study demonstrated that the sugar metabolism of fibroblast-like synoviocytes was significantly disturbed by RA [85]. Kim et al. also pointed out that the sugar metabolism level in RA is higher than healthy people and higher than osteoarthritis, which makes it a distinct feature for RA [85]. As presented in the previous chapter, a high glycolysis level is a characteristic of osteoclastogenesis from monocytic progenitors with an increased glycolytic genes expression [17]. This switch of energy consumption can explain the tendency towards glycolysis and mitochondrial defect in RA. The symptoms of arthritis are caused by the destroyed cartilage; consequently, the abnormal enhancement of osteoclast and/or immune cell activity by fatty acids or glycolysis level is closely related with the disease conditions.
3.4. Osteosarcoma
As the most common histological form of the bone cancerous tumor, osteosarcoma causes severe symptoms by malignant neoplasia, threatening people of all ages [86][87]. A clear presentation of metabolic heterogeneity in oncogenesis and metastasis benefits early diagnosis and enhances the understanding of the clinical behavior of tumor tissue. Among all the pathologies, osteoblastic tumor holds the highest proportion, followed by chondroblastic and fibroblastic [86][88]. Several studies have been performed on osteosarcoma with a focus on abnormal metabolisms. An NMR profiling on blood serum samples revealed that energy metabolism was enormously altered throughout tumorigenesis [86]. The abnormal levels of amino acids, fatty acids, and glucose were found, suggesting an alternative energy source for cancer cells. In most cancer cases, glycolysis is considered to be the primary energy source for tumor tissue, while oxidative phosphorylation level is decreased [89]. A similar decrease in TCA cycle metabolite levels was detected in an in vitro experiment using osteosarcoma stem cells [90]. This preference for energic metabolism was verified by metabolite enrichment analysis, of which metabolite biomarkers involved in the glycolysis pathway were highly evaluated [90][91]. The lower TCA cycle level in osteosarcoma tumor tissue was explained by the down-regulation of mitochondrial function, accompanied by a reduction of glutamate, aspartate, and glutathione with an elevation of Gln [90][92]. The mitochondrial dysfunction caused by the expression of metastasis genes in osteosarcoma cells resulted in metabolic disorder and presented an aggressive phenotype [93].
The metabolite-based osteosarcoma biomarkers can be potentially employed not only for diagnosis but also to monitor disease progression. These metabolites can be attributed to the development of regulators for energy metabolism and cellular stress in osteosarcoma tumor tissue. Besides, targeting metabolic pathways provides new therapeutic strategies. For example, new osteosarcoma therapy targeting the amino acid metabolism of cancer stem cells has been suggested [90].
3.5. Summary of Metabolic Pathways Relevant in Bone Diseases
To comprehensively identify metabolic pathways that are affected during bones diseases, we conducted an integrating enrichment and pathway topology analysis from the data of Table 1 by using Metabo Analyst 5.0 [94]. For this, we used the metabolites that are reported as significantly altered in osteoporosis, arthritis, and osteosarcoma.
The metabolic pathways prominently altered in both osteoporosis and arthritis are (1) aminoacyl-tRNA biosynthesis, (2) arginine and proline metabolism, (3) arginine biosynthesis, and (4) valine, leucine and isoleucine biosynthesis. At the same time, the most altered pathways from osteosarcoma are (1) alanine, aspartate and glutamate metabolism, (2) TCA cycle, and (3) arginine biosynthesis. Interestingly, a high degree of similarity of the most altered metabolic pathways has been found between osteoporosis and arthritis. This phenomenon may be caused by the pathophysiological similarity of these two diseases: abnormal resorption of the target tissue by osteoclasts, macrophages and/or some other immune cells. However, due to the limitation of the data sources and the diversity of various original studies, this conclusion needs further research and discussion. Most notably, arginine synthesis and metabolism are remarkably altered in all mentioned diseases. The metabolism of glutamate, a precursor of arginine, is also involved in OS. The immune system, therefore, plays a critical role in bone diseases as a regulator of immune responses and arginine, and its related metabolic pathways have great value for the future bone research [95]. It should be pointed out that metabolic pathways altered in bone diseases have been previously reported to be relevant in other diseases. The control of physiopathological processes such as angiogenesis, inflammation, and tumorigenesis is related to aminoacyl-tRNA synthesis [96]. Arginine and proline have important roles in wound healing, antioxidative reactions, and immune responses, as a result, the metabolites’ variation after tissue injury is rational [97]. Obesity and cancer risk are also linked with proline metabolism, leading to various of complications [98]. The alteration of glycolysis, TCA cycle and glutaminolysis are typical signals in cancers, [99][100]. To conclude, some of the changes in metabolism observed in bone diseases reflect more general pathophysiological processes such as inflammation. Further studies are needed to elucidate how bone diseases may influence or be influenced by other diseases under a certain physical condition of the patients.
Table 1. Overview of reported metabolite changes in the different diseases, used analytical methods and corresponding references.
| Ref. | Disease | Technique | Sample Type | Metabolite Changes |
|---|---|---|---|---|
| [46] | POP | GC/TOF-MS | Rat plasma | ↑ Arachidonic acid, octadecadienoic acid, valine, leucine, isoleucine, homocysteine, hydroxyproline, ketone bodies ↓ Docosahexaenoic acid, dodecanoic acid, lysine |
| [53] | POP | UPLC/Q-TOF-MS | Rat bone tissue | ↑ Lysophosphatidylcholine, phosphatidylcholine, ceramide, phosphoserine ↓ Uridine, hypoxanthine, xanthine, inosine, cytidine, phenylalanine, leucine, carnitine, proline, arginine |
| [101] | POP | 1H NMR | Rat urine | ↑ Trigonelline, phosphocreatine, pyruvate, methylamine, trimethylamine oxide ↓ Benzoic acid, dimethylamine, trimethylamine, threonic acid, alanine, leucine, 2-ketoglutarate, allantoin, acetate, formate |
| [47] | POP | GC/TOF-MS | Rat plasma | ↑ Arachidonic acid, homocysteine, homocysteine, ethanedioic acid↓ Alanine, malic acid, citric acid, fructose involved |
| [45] | POP | GC-MS | Women serum | ↑ Arachidonic acid, lysine, eicosadienoic acid, oleic acid, linoleic acid, allose, tryptophan ↓ Homoserine, 3-hydroxy-l-proline, pyruvic acid |
| [102] | POP | UPLC-Q-TOF/MS | Rat serum | ↑ Lysine, linoleic acid, hippuric acid, octadecadienoic acid, carnitine, glucose, arginine, S-adenosylhomocysteine, ornithine, tryptophan, arachidonic acid, methionyl-hydroxyproline ↓ Homoserine, 3-hydroxy-l-proline, pyruvic acid, citric acid, estriol, 8-HETE, uric acid, glutamine, glyceraldehyde, palmitic acid, 4-oxoretinol, taurocholic acid |
| [103] | DOP | NMR | Human plasma | ↑ Leucine, isoleucine valine, alanine, N-acetylglycoprotein, inositol, proline, glucose, glutamine, 1-methyl-histidine, tyrosine ↓ O-acetylglycoprotein, α-ketoglutaric acid, citrate, creatine |
| [83] | RA | NMR | Human synovial | ↑ Threonine, xanthine, methylsuccinate, glutamate, methylmalonate, taurine, lactate, pyruvate, propylene glycol, leucine, tyrosine, 3-hydroxybutyrate ↓ Creatinine, creatine, o-acetyl carnitine, L-carnitine, betaine, choline, formate, glycine, asparagine, formate, acetate, phenylalanine, succinate, pantothenate, fumarate, acetoacetate, acetone, lysine |
| [85] | RA | GC/TOF-MS | Human synoviocytes | ↑ Inosine, urate, guanine, behenic acid, palmitoleic acid, arachidic acid, oleic acid, glucose-6-phosphate, phosphogluconic acid, aspartate, adipate, asparagine ↓ Isoleucine, leucine, leucine, histidine, valine, ornithine, lysine, methionine sulfoxide, tryptophan, mannitol, xylose |
| [104] | RA | LCMS | Human plasma | ↑ Kynurenine, indolelactic acid, hypoxanthine, cholesterol, triglyceride, lysophospatidylcholines ↓ Tryptophan, fatty acids, acylcarnitines |
| [105] | OA | MALDI-MSI | Human bone marrow MSCs | ↑ Arachidonic acid, oleic acid, stearic acid, dihydroxyacetone phosphate, phosphatidylglycerol, phosphatidylinositol ↓ Myoinositol, phosphatidic acid, lysophosphatidic acid, glutamine |
| [90] | OS | UHPLC-QE-MS | Human osteosarcoma stem cells | ↑ Aspartic acid, asparagine, glutamine, arginine, ornithine, methionine, methylthioadenosine ↓ succinic acid, citric acid, aconitic acid, oxoglutaric acid, ureidosuccinic acid |
| [106] | OS | UHPLC-HRMS | Human serum | ↑ Adenosine monophosphate, inosinic acid, guanosine monophosphate, hypoxanthine, lactic acid ↓ Uric acid, 4-hydroxybenzoic acid, testosterone sulfate, iminodiacetic acid, 3-carboxy-4-methyl-5-propyl-2-furanpropionic acid, decanoylcarnitine |
| [107] | OS | GC-MS | Human serum and urine | ↑ Cystine, 2-hydroxybutyrate, inosine, creatinine, putrescine, aspartate, proline, galactopyranose ↓ Malate, fumarate, pyruvate, lactate, sucrose, dodecanoate, glycerol phosphate, creatinine |
| [62] | Bone Injuries | UPLC-MS/MS | Trauma patient plasma | ↑ Myristoleate, hexadecadienoate, octadecadienedioate, choline phosphate, phosphoethanolamine, pregnenolone sulfate, cortisol, glycerol 3-phosphate, beta-citrylglutamate, trans-urocanate, kynurenate, cysteine, spermidine, cysteinyl glycine ↓ Decanoyl carnitine, 2-hydroxyheptanoate, 4-hydroxynonenal, glycerophosphoethanolamine, palmitoyl-linoleoyl-glycerol, stearoyl-linoleoyl-glycerol, cholate, n-acetylglutamine, pyroglutamine, tryptophan, cysteine s-sulfate |
| [108] | Bone Injuries | LC-MS/MS | Human bone marrow plasma | ↑ Kynurenine |
References
- Kemmak, A.R.; Rezapour, A.; Jahangiri, R.; Nikjoo, S.; Farabi, H.; Soleimanpour, S. Economic Burden of Osteoporosis in the World: A Systematic Review. Med. J. Islam. Repub. Iran. 2020, 2020, 1–8.
- Wade, S.W.; Strader, C.; Fitzpatrick, L.A.; Anthony, M.S.; O’Malley, C.D. Estimating prevalence of osteoporosis: Examples from industrialized countries. Arch. Osteoporos. 2014, 9, 182.
- Tian, L.; Yang, R.; Wei, L.; Liu, J.; Yang, Y.; Shao, F.; Ma, W.; Li, T.; Wang, Y.; Guo, T. Prevalence of osteoporosis and related lifestyle and metabolic factors of postmenopausal women and elderly men. Medicine 2017, 96.
- Guijas, C.; Montenegro-Burke, J.R.; Warth, B.; Spilker, M.E.; Siuzdak, G. Metabolomics activity screening for identifying metabolites that modulate phenotype. Nat. Biotechnol. 2018, 36, 316–320.
- Armiento, A.R.; Hatt, L.P.; Sanchez Rosenberg, G.; Thompson, K.; Stoddart, M.J. Functional Biomaterials for Bone Regeneration: A Lesson in Complex Biology. Adv. Funct. Mater. 2020, 30, 1–41.
- Ma, C.; Kuzma, M.L.; Bai, X.; Yang, J. Biomaterial-Based Metabolic Regulation in Regenerative Engineering. Adv. Sci. 2019, 6.
- Kubatzky, K.F.; Uhle, F.; Eigenbrod, T. From macrophage to osteoclast—How metabolism determines function and activity. Cytokine 2018, 112, 102–115.
- Liang, S.T.; Chen, J.R.; Tsai, J.J.; Lai, Y.H.; Hsiao, C. Der Overexpression of notch signaling induces hyperosteogeny in zebrafish. Int. J. Mol. Sci. 2019, 20, 3613.
- Mohammadalipour, A.; Dumbali, S.P.; Wenzel, P.L. Mitochondrial Transfer and Regulators of Mesenchymal Stromal Cell Function and Therapeutic Efficacy. Front. Cell Dev. Biol. 2020, 8, 1–22.
- Guntur, A.R.; Le, P.T.; Farber, C.R.; Rosen, C.J. Bioenergetics during calvarial osteoblast differentiation reflect strain differences in bone mass. Endocrinology 2014, 155, 1589–1595.
- Yao, Y.; Deng, Q.; Song, W.; Zhang, H.; Li, Y.; Yang, Y.; Fan, X.; Liu, M.; Shang, J.; Sun, C.; et al. MIF Plays a Key Role in Regulating Tissue-Specific Chondro-Osteogenic Differentiation Fate of Human Cartilage Endplate Stem Cells under Hypoxia. Stem Cell Rep. 2016, 7, 249–262.
- Shum, L.C.; White, N.S.; Mills, B.N.; De Mesy Bentley, K.L.; Eliseev, R.A. Energy Metabolism in Mesenchymal Stem Cells during Osteogenic Differentiation. Stem Cells Dev. 2016, 25, 114–122.
- Pattappa, G.; Heywood, H.K.; de Bruijn, J.D.; Lee, D.A. The metabolism of human mesenchymal stem cells during proliferation and differentiation. J. Cell. Physiol. 2011, 226, 2562–2570.
- Lee, W.C.; Ji, X.; Nissim, I.; Long, F. Malic Enzyme Couples Mitochondria with Aerobic Glycolysis in Osteoblasts. Cell Rep. 2020, 32, 108108.
- Lee, S.Y.; Abel, E.D.; Long, F. Glucose metabolism induced by Bmp signaling is essential for murine skeletal development. Nat. Commun. 2018, 9, 1–11.
- Kim, J.; Jeong, D.; Kang, H.K.; Jung, S.Y.; Kang, S.S.; Min, B. Cellular Physiology and Biochemistr y Biochemistry Osteoclast Precursors Display Dynamic Metabolic Shifts toward Accelerated Glucose Metabolism at an Early Stage of RANKL-Stimulated Osteoclast Differentiation. Cell Physiol. Biochem. 2007, 749, 935–946.
- Indo, Y.; Takeshita, S.; Ishii, K.A.; Hoshii, T.; Aburatani, H.; Hirao, A.; Ikeda, K. Metabolic regulation of osteoclast differentiation and function. J. Bone Miner. Res. 2013, 28, 2392–2399.
- Arnett, T.R.; Orriss, I.R. Metabolic properties of the osteoclast. Bone 2018, 115, 25–30.
- McDonald, M.M.; Khoo, W.H.; Ng, P.Y.; Xiao, Y.; Zamerli, J.; Thatcher, P.; Kyaw, W.; Pathmanandavel, K.; Grootveld, A.K.; Moran, I.; et al. Osteoclasts recycle via osteomorphs during RANKL-stimulated bone resorption. Cell 2021, 184, 1330–1347.
- Manolagas, S.C. Birth and death of bone cells: Basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr. Rev. 2000, 21, 115–137.
- Jacome-Galarza, C.E.; Percin, G.I.; Muller, J.T.; Mass, E.; Lazarov, T.; Eitler, J.; Rauner, M.; Yadav, V.K.; Crozet, L.; Bohm, M.; et al. Developmental origin, functional maintenance and genetic rescue of osteoclasts. Nature 2019, 568, 541–545.
- Kasonga, A.E.; Deepak, V.; Kruger, M.C.; Coetzee, M. Arachidonic Acid and Docosahexaenoic Acid Suppress Osteoclast Formation and Activity in Human CD14 + Monocytes, In vitro. PLoS ONE 2015, 1–19.
- Ryu, J.; Kim, H.; Chang, E.J.; Kim, H.J.; Lee, Y.; Kim, H.H. Proteomic analysis of osteoclast lipid rafts: The role of the integrity of lipid rafts on V-ATPase activity in osteoclasts. J. Bone Miner. Metab. 2010, 28, 410–417.
- Liu, X.; Liu, Y.; Cheng, M.; Zhang, X.; Xiao, H. A metabolomics study of the inhibitory effect of 17-beta-estradiol on osteoclast proliferation and differentiatio. Mol. Biosyst. 2015, 11, 635–646.
- Van Gastel, N.; Stegen, S.; Eelen, G.; Schoors, S. Lipid availability determines skeletal progenitor cell fate via SOX9. Nature 2020, 579, 111–117.
- Dar, H.Y.; Azam, Z.; Anupam, R.; Mondal, R.K.; Srivastava, R.K. Osteoimmunology: The Nexus between bone and immune system. Front. Biosci. Landmark 2018, 23, 464–492.
- Rath, M.; Müller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via arginase or nitric oxide synthase: Two competing arginine pathways in macrophages. Front. Immunol. 2014, 5.
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The metabolic signature of macrophage responses. Front. Immunol. 2019, 10, 1–16.
- Rodriguez, P.C.; Ochoa, A.C.; Al-Khami, A.A. Arginine metabolism in myeloid cells shapes innate and adaptive immunity. Front. Immunol. 2017, 8.
- Climaco-Arvizu, S.; Domínguez-Acosta, O.; Cabañas-Cortés, M.A.; Rodríguez-Sosa, M.; Gonzalez, F.J.; Vega, L.; Elizondo, G. Aryl hydrocarbon receptor influences nitric oxide and arginine production and alters M1/M2 macrophage polarization. Life Sci. 2016, 155, 76–84.
- Chen, Z.; Klein, T.; Murray, R.Z.; Crawford, R.; Chang, J.; Wu, C.; Xiao, Y. Osteoimmunomodulation for the development of advanced bone biomaterials. Mater. Today 2016, 19, 304–321.
- Lu, L.Y.; Loi, F.; Nathan, K.; Lin, T.H.; Pajarinen, J.; Gibon, E.; Nabeshima, A.; Cordova, L.; Jämsen, E.; Yao, Z.; et al. Pro-inflammatory M1 macrophages promote Osteogenesis by mesenchymal stem cells via the COX-2-prostaglandin E2 pathway. J. Orthop. Res. 2017, 35, 2378–2385.
- Wang, F.; Zhang, S.; Vuckovic, I.; Jeon, R.; Lerman, A.; Folmes, C.D.; Dzeja, P.P.; Herrmann, J. Glycolytic Stimulation Is Not a Requirement for M2 Macrophage Differentiation. Cell Metab. 2018, 28, 463–475.
- Jha, A.K.; Huang, S.C.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430.
- Muñoz, J.; Akhavan, N.S.; Mullins, A.P.; Arjmandi, B.H. Macrophage polarization and osteoporosis: A review. Nutrients 2020, 12, 2999.
- Lv, H.; Jiang, F.; Guan, D.; Lu, C.; Guo, B.; Chan, C.; Peng, S.; Liu, B.; Guo, W.; Zhu, H.; et al. Metabolomics and Its Application in the Development of Discovering Biomarkers for Osteoporosis Research. Int. J. Mol. Sci. 2018, 17, 2018.
- Zhang, A.h.; Ma, Z.m.; Sun, H.; Zhang, Y.; Liu, J.h.; Wu, F.f.; Wang, X.j. High-throughput metabolomics evaluate the efficacy of total lignans from Acanthophanax senticosus stem against ovariectomized osteoporosis rat. Front. Pharmacol. 2019, 10.
- Jiang, Y.; Li, Y.; Zhou, L.; Zhang, D. UPLC-MS metabolomics method provides valuable insights into the effect and underlying mechanisms of Rhizoma Drynariae protecting osteoporosis. J. Chromatogr. B 2020, 1152, 122262.
- Longo, A.B.; Ward, W.E. PUFAs, bone mineral density, and fragility fracture: Findings from human studies. Adv. Nutr. 2016, 7, 299–312.
- Xia, T.; Dong, X.; Lin, L.; Jiang, Y.; Ma, X.; Xin, H.; Zhang, Q.; Qin, L. Metabolomics profiling provides valuable insights into the underlying mechanisms of Morinda officinalis on protecting glucocorticoid-induced osteoporosis. J. Pharm. Biomed. Anal. 2019, 166, 336–346.
- Lee, N.K.; Choi, Y.G.; Baik, J.Y.; Han, S.Y.; Jeong, D.W.; Bae, Y.S.; Kim, N.; Lee, S.Y. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood 2005, 106, 852–859.
- Kim, H.J.; Ohk, B.; Yoon, H.J.; Kang, W.Y.; Seong, S.J.; Kim, S.Y.; Yoon, Y.R. Docosahexaenoic acid signaling attenuates the proliferation and differentiation of bone marrow-derived osteoclast precursors and promotes apoptosis in mature osteoclasts. Cell. Signal. 2017, 29, 226–232.
- Liu, X.H.; Kirschenbaum, A.; Weinstein, B.M.; Zaidi, M.; Yao, S.; Levine, A.C. Prostaglandin E2 modulates components of the Wnt signaling system in bone and prostate cancer cells. Biochem. Biophys. Res. Commun. 2010, 394, 715–720.
- Garcia, C.; Boyce, B.F.; Gilles, J.; Dallas, M.; Qiao, M.E.; Mundy, G.R.; Bonewald, L.F. Leukotriene B, Stimulates Osteoclastic Bone Resorption. J. Bone Miner. Res. 1996, 1619–1627.
- Qi, H.; Bao, J.; An, G.; Ouyang, G.; Zhang, P.; Wang, C.; Ying, H.; Ouyang, P.; Ma, B.; Zhang, Q. Association between the metabolome and bone mineral density in pre- and post-menopausal Chinese women using GC-MS. Mol. Biosyst. 2016, 12, 2265–2275.
- Ma, B.; Liu, J.; Zhang, Q.; Ying, H.; Jiye, A.; Sun, J.; Wu, D.; Wang, Y.; Li, J.; Liu, Y. Metabolomic Profiles Delineate Signature Metabolic Shifts during Estrogen Deficiency-Induced Bone Loss in Rat by GC-TOF/MS. PLoS ONE 2013, 8, 1–10.
- Ma, B.; Li, X.; Zhang, Q.; Wu, D.; Wang, G.; Jiye, A.; Sun, J.; Li, J.; Liu, Y.; Wang, Y.; et al. Metabonomic profiling in studying anti-osteoporosis effects of strontium fructose 1,6-diphosphate on estrogen deficiency-induced osteoporosis in rats by GC/TOF-MS. Eur. J. Pharmacol. 2013, 718, 524–532.
- Almeida, M.; Ambrogini, E.; Han, L.; Manolagas, S.C.; Jilka, R.L. Increased lipid oxidation causes oxidative stress, increased peroxisome proliferator-activated receptor-γ expression, and diminished pro-osteogenic Wnt signaling in the skeleton. J. Biol. Chem. 2009, 284, 27438–27448.
- Kawai, M.; Rosen, C.J. PPARγ: A circadian transcription factor in adipogenesis and osteogenesis. Nat. Rev. Endocrinol. 2010, 6, 629–636.
- Li, Y.; Jin, D.; Xie, W.; Wen, L.; Chen, W.; Xu, J.; Ding, J.; Ren, D. PPAR-γ and Wnt Regulate the Differentiation of MSCs into Adipocytes and Osteoblasts Respectively. Curr. Stem Cell Res. Ther. 2017, 13, 185–192.
- Casado-Díaz, A.; Santiago-Mora, R.; Dorado, G.; Quesada-Gómez, J.M. The omega-6 arachidonic fatty acid, but not the omega-3 fatty acids, inhibits osteoblastogenesis and induces adipogenesis of human mesenchymal stem cells: Potential implication in osteoporosis. Osteoporos. Int. 2013, 24, 1647–1661.
- Coetzee, M.; Haag, M.; Kruger, M.C. Effects of arachidonic acid, docosahexaenoic acid, prostaglandin E2 and parathyroid hormone on osteoprotegerin and RANKL secretion by MC3T3-E1 osteoblast-like cells. J. Nutr. Biochem. 2007, 18, 54–63.
- Zhao, H.; Li, X.; Zhang, D.; Chen, H.; Chao, Y.; Wu, K.; Dong, X.; Su, J. Integrative Bone Metabolomics—Lipidomics Strategy for Pathological Mechanism of Postmenopausal Osteoporosis Mouse Model. Sci. Rep. 2018, 8, 1–11.
- Choi, Y.J.; Song, I.; Jin, Y.; Jin, H.S.; Ji, H.M.; Jeong, S.Y.; Won, Y.Y.; Chung, Y.S. Transcriptional profiling of human femoral mesenchymal stem cells in osteoporosis and its association with adipogenesis. Gene 2017, 632, 7–15.
- Duque, G.; Troen, B.R. Understanding the mechanisms of senile osteoporosis: New facts for a major geriatric syndrome. J. Am. Geriatr. Soc. 2008, 56, 935–941.
- Abuna, R.P.F.; Almeida, L.O.; Souza, A.T.P.; Fernandes, R.R.; Sverzut, T.F.V.; Rosa, A.L.; Beloti, M.M. Osteoporosis and osteoblasts cocultured with adipocytes inhibit osteoblast differentiation by downregulating histone acetylation. J. Cell. Physiol. 2020, 1–12.
- Wu, Y.; Zhang, M.; Chen, X.; Zhou, Y.; Chen, Z. Metabolomic analysis to elucidate the change of the n-3 polyunsaturated fatty acids in senescent osteoblasts. Biosci. Biotechnol. Biochem. 2021, 85, 611–620.
- Zhang, X.; Xu, H.; Li, G.H.Y.; Long, M.T.; Cheung, C.; Vasan, R.S.; Hsu, Y.; Kiel, D.P.; Liu, C. Metabolomics insights into osteoporosis through association with bone mineral density. J. Bone Miner. Res. 2021, 36, 729–738.
- Kuo, T.R.; Chen, C.H. Bone biomarker for the clinical assessment of osteoporosis: Recent developments and future perspectives. Biomark. Res. 2017, 5, 5–13.
- Miyamoto, T.; Hirayama, A.; Sato, Y.; Koboyashi, T.; Katsuyama, E.; Kanagawa, H.; Miyamoto, H.; Mori, T.; Yoshida, S.; Fujie, A.; et al. A serum metabolomics-based profile in low bone mineral density postmenopausal women. Bone 2017, 95, 1–4.
- Jagtap, V.R.; Ganu, J.V. Effect of antiresorptive therapy on urinary hydroxyproline in postmenopausal osteoporosis. Indian J. Clin. Biochem. 2012, 27, 90–93.
- Ibrahim, H.; Alnachoukati, O.; Baxter, B.A.; Chapin, T.; Schroeppel, T.; Dunn, J.; Ryan, E.P. Non-Targeted Metabolomics Signature in the Plasma and Bone Marrow of Patients with Long Bone Injuries. Curr. Metab. Syst. Biol. 2019, 7, 51–66.
- Calder, P.C. Fatty acids: Long-chain fatty acids and inflammation. Proc. Nutr. Soc. 2012, 71, 284–289.
- Ghezzi, P. Role of glutathione in immunity and inflammation in the lung. Int. J. Gen. Med. 2011, 4, 105–113.
- Kayoko, S.; Chian, J.J.; Kyoko, T.; Schaffer, S.W. Role of ROS Production and Turnover in the Antioxidant Activity of Taurine. Adv. Exp. Med. Biol. 2015, 803.
- Poli, G.; Schaur, R.J. 4-Hydroxynonenal in the pathomechanisms of oxidative stress. IUBMB Life 2000, 50, 315–321.
- Almadi, T.; Cathers, I.; Chow, C.M. Associations among work-related stress, cortisol, inflammation, and metabolic syndrome. Psychophysiology 2013, 50, 821–830.
- Dinçel, E.; Özkan, Y.; Şüküroğlu, M.; Özsoy, H.; Sepici Dinçel, A. Evaluation of tryptophan/kynurenine pathway relevance with immune system biomarkers of low energy trauma hip fractures in osteoporotic patients. Arch. Rheumatol. 2017, 32, 203–208.
- Chen, Y.; Guillemin, G.J. Kynurenine Pathway Metabolites in Humans: Disease and Healthy States. Int. J. Tryptophan. Res. 2010, 61, 1–19.
- Wolf, A.M.; Wolf, D.; Rumpold, H.; Moschen, A.R.; Kaser, A.; Obrist, P.; Fuchs, D.; Brandacher, G.; Winkler, C.; Geboes, K.; et al. Overexpression of indoleamine 2,3-dioxygenase in human inflammatory bowel disease. Clin. Immunol. 2004, 113, 47–55.
- Jasperson, L.K.; Bucher, C.; Panoskaltsis-Mortari, A.; Taylor, P.A.; Mellor, A.L.; Munn, D.H.; Blazar, B.R. Indoleamine 2,3-dioxygenase is a critical regulator of acute graft-versus-host disease lethality. Blood 2008, 111, 3257–3265.
- Westbrook, A.M.; Wei, B.; Hacke, K.; Xia, M.; Braun, J.; Schiestl, R.H. The role of tumour necrosis factor-α and tumour necrosis factor receptor signalling in inflammation-associated systemic genotoxicity. Mutagenesis 2012, 27, 77–86.
- Yoon, B.-H.; Yu, W. Clinical Utility of Biochemical Marker of Bone Turnover: Fracture Risk Prediction and Bone Healing. J. Bone Metab. 2018, 25, 73.
- Veitch, S.W.; Findlay, S.C.; Hamer, A.J.; Blumsohn, A.; Eastell, R.; Ingle, B.M. Changes in bone mass and bone turnover following tibial shaft fracture. Osteoporos. Int. 2006, 17, 364–372.
- Sousa, C.P.; Lopez-Peña, M.; Guzón, F.M.; Abreu, H.V.D.; Luís, M.R.; Viegas, C.A.; Camassa, J.; Azevedo, J.T.D.; Cabrita, A.S.; Reis, R.L.; et al. Evaluation of bone turnover markers and serum minerals variations for predicting fracture healing versus non-union processes in adult sheep as a model for orthopedic research. Injury 2017, 48, 1768–1775.
- McInnes, I.B.; Schett, G. The Pathogenesis of Rheumatoid Arthritis. N. Engl. J. Med. 2011, 365, 2205–2219.
- Innes, J.K.; Calder, P.C. Omega-6 fatty acids and inflammation. Prostaglandins Leukot. Essent. Fat. Acids 2018, 132, 41–48.
- Calder, P.C. Fatty acids and inflammation: The cutting edge between food and pharma. Eur. J. Pharmacol. 2011, 668, S50–S58.
- Miles, E.A.; Calder, P.C. Influence of marine n-3 polyunsaturated fatty acids on immune function and a systematic review of their effects on clinical outcomes in rheumatoid arthritis. Br. J. Nutr. 2012, 107.
- Rosser, E.C.; Piper, C.J.M.; Matei, D.E.; Blair, P.A.; Rendeiro, A.F.; Orford, M.; Alber, D.G.; Krausgruber, T.; Catalan, D.; Klein, N.; et al. Microbiota-Derived Metabolites Suppress Arthritis by Amplifying Aryl-Hydrocarbon Receptor Activation in Regulatory B Cells. Cell Metab. 2020, 31, 837–851.
- Mizuno, M.; Noto, D.; Kaga, N.; Chiba, A.; Miyake, S. The dual role of short fatty acid chains in the pathogenesis of autoimmune disease models. PLoS ONE 2017, 12, 1–15.
- Hui, W.; Yu, D.; Cao, Z.; Zhao, X. Butyrate inhibit collagen-induced arthritis via Treg/IL-10/Th17 axis. Int. Immunopharmacol. 2019, 68, 226–233.
- Narasimhan, R.; Coras, R.; Rosenthal, S.B.; Sweeney, S.R.; Lodi, A.; Tiziani, S.; Boyle, D.; Kavanaugh, A.; Guma, M. Serum metabolomic profiling predicts synovial gene expression in rheumatoid arthritis. Arthritis Res. Ther. 2018, 20, 1–11.
- Weyand, C.M.; Wu, B.; Goronzy, J.J. The metabolic signature of T cells in rheumatoid arthritis. Curr. Opin. Rheumatol. 2020, 32, 159–167.
- Kyong, J.; Kim, S.; Hwang, J.; Kim, J.; Heon, K.; Cha, H. Original article GC / TOF-MS-based metabolomic profiling in cultured fibroblast-like synoviocytes from rheumatoid arthritis. Jt. Bone Spine 2016, 83, 707–713.
- Quintero Escobar, M.; Costa, T.B.B.C.; Martins, L.G.; Costa, S.S.; van Helvoort Lengert, A.; Boldrini, E.; Morini da Silva, S.R.; Lopes, L.F.; Vidal, D.O.; Krepischi, A.C.V.; et al. Insights in Osteosarcoma by Proton Nuclear Magnetic Resonance Serum Metabonomics. Front. Oncol. 2020, 10, 1–9.
- Tandon, R. Future directions in the treatment of schizophrenia. Pharmacol. Treat. Schizophr. 2012, 67–79.
- Hauben, E.I.; Weeden, S.; Pringle, J.; Van Marck, E.A. Does the histological subtype of high-grade central osteosarcoma influence the response to treatment with chemotherapy and does it affect overall survival ? A study on 570 patients of two consecutive trials of the European Osteosarcoma Intergroup. Eur. J. Cancer 2002, 38, 1218–1225.
- Song, J.; Wu, X.; Liu, F.; Li, M.; Sun, Y.; Wang, Y.; Wang, C.; Zhu, K.; Jia, X.; Wang, B.; et al. Long non-coding RNA PVT1 promotes glycolysis and tumor progression by regulating miR-497/HK2 axis in osteosarcoma. Biochem. Biophys. Res. Commun. 2017, 490, 217–224.
- Zhong, Z.; Mao, S.; Lin, H.; Li, H.; Lin, J.; Lin, J.M. Alteration of intracellular metabolome in osteosarcoma stem cells revealed by liquid chromatography-tandem mass spectrometry. Talanta 2019, 204, 6–12.
- Chen, K.; Zhu, C.; Cai, M.; Fu, D. Integrative metabolome and transcriptome profiling reveals discordant glycolysis process between osteosarcoma and normal osteoblastic cells. J. Cancer Res. Clin. 2014.
- Ren, L.; Dowdy, T.; Huang, S.; Issaq, S.H.; Beck, J.; Wang, H.; Hoang, C.T.; Lita, A.; Larion, M.; Leblanc, A.K. Glutaminase-1 (GLS1) inhibition limits metastatic progression in osteosarcoma. Cancer Metab. 2020, 8, 1–13.
- Jackson, M.; Serada, N.; Sheehan, M.; Srinivasan, S.; Mason, N.; Id, M.G.; Avadhani, N. Mitochondrial genome and functional defects in osteosarcoma are associated with their aggressive phenotype. PLoS ONE 2018, 13, 1–20.
- Pang, Z.; Chong, J.; Zhou, G.; Anderson, D.; Morais, D.L.; Chang, L.; Barrette, M.; Gauthier, C.; Etienne, P.; Li, S.; et al. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 1–9.
- Bronte, V.; Zanovello, P. Regulation of immune responses by L-arginine metabolism. Nat. Rev. Immunol. 2005, 5, 641–654.
- Yao, P.; Fox, P.L. Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol. Med. 2013, 5, 332–343.
- Wu, Y.; Streijger, F.; Wang, Y.; Lin, G.; Christie, S.; Mac-Thiong, J.M.; Parent, S.; Bailey, C.S.; Paquette, S.; Boyd, M.C.; et al. Parallel Metabolomic Profiling of Cerebrospinal Fluid and Serum for Identifying Biomarkers of Injury Severity after Acute Human Spinal Cord Injury. Sci. Rep. 2016, 6, 1–14.
- Phang, J.M.; Liu, W.; Zabirnyk, O. Proline metabolism and microenvironmental stress. Annu. Rev. Nutr. 2010, 30, 441–463.
- Ganapathy-kanniappan, S.; Geschwind, J.-F. Tumor glycolysis as a target for cancer therapy. Mol. Cancer 2013, 12, 1–11.
- Chen, J.Q.; Russo, J. Dysregulation of glucose transport, glycolysis, TCA cycle and glutaminolysis by oncogenes and tumor suppressors in cancer cells. Biochim. Biophys. Acta Rev. Cancer 2012, 1826, 370–384.
- Liu, S.; Yuan, X.; Ma, C.; Zhao, J.; Xiong, Z. 1H-NMR-based urinary metabolomic analysis for the preventive effects of gushudan on glucocorticoid-induced osteoporosis rats. Anal. Biochem. 2020, 610, 113992.
- Si, Z.; Zhou, S.; Shen, Z.; Luan, F. High-Throughput Metabolomics Discovers Metabolic Biomarkers and Pathways to Evaluating the Efficacy and Exploring Potential Mechanisms of Osthole Against Osteoporosis Based on UPLC/Q-TOF-MS Coupled With Multivariate Data Analysis. Front. Pharmacol. 2020, 11, 1–15.
- Liang, W.D.; Huang, P.J.; Xiong, L.H.; Zhou, S.; Ye, R.Y.; Liu, J.R.; Wei, H.; Lai, R.Y. Metabolomics and its application in the mechanism analysis on diabetic bone metabolic abnormality. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 9591–9600.
- Rantapa, S.; Surowiec, I.; Lisbeth, A. Metabolite and Lipid Profiling of Biobank Plasma Samples Collected Prior to Onset of Rheumatoid Arthritis. PLoS ONE 2016, 1–14.
- Rocha, B.; Cillero-pastor, B.; Eijkel, G.; Calamia, V.; Fernandez-puente, P.; Paine, M.R.L.; Ruiz-romero, C.; Heeren, R.M.A.; Blanco, F.J.; Rocha, B.; et al. Integrative Metabolic Pathway Analysis Reveals Novel Therapeutic Targets in Osteoarthritis Authors Integrative Metabolic Pathway Analysis. J. Title Mol. Cell. Proteomics 2020, 19, 574–588.
- Lv, D.; Zou, Y.; Zeng, Z.; Yao, H.; Ding, S.; Bian, Y.; Wen, L.; Xie, X. Comprehensive metabolomic profiling of osteosarcoma based on UHPLC-HRMS. Metabolomics 2020, 16, 1–11.
- Zhang, Z.; Qiu, Y.; Hua, Y.; Wang, Y.; Chen, T.; Zhao, A.; Chi, Y.; Pan, L.; Hu, S.; Li, J.; et al. Serum and urinary metabonomic study of human osteosarcoma. J. Proteome Res. 2010, 9, 4861–4868.
- Le, A.R.T.I.C.; Metabolism, B.; Kim, B.; Hamrick, M.W.; Yoo, H.J.; Lee, S.H.; Kim, S.J.; Koh, J.; Isales, C.M.; Kim, O.B. The Detrimental Effects of Kynurenine, a Tryptophan Metabolite, on Human Bone Metabolism. J. Clin. Endocrinol. Metab. 2019, 104, 2334–2342.
More
Information
Subjects:
Endocrinology & Metabolism; Orthopedics
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
3 times
(View History)
Update Date:
28 Sep 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No