 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Myriam MERARCHI | + 1477 word(s) | 1477 | 2021-07-07 06:08:53 | | | |
| 2 | Vivi Li | Meta information modification | 1477 | 2021-07-07 11:30:59 | | |
Video Upload Options
For the last couple of decades, natural products, either applied singly or in conjunction with other cancer therapies including chemotherapy and radiotherapy, have allowed us to combat different types of human cancers through the inhibition of their initiation and progression. The principal sources of these useful compounds are isolated from plants that were described in traditional medicines for their curative potential. Leelamine, derived from the bark of pine trees, was previously reported as having a weak agonistic effect on cannabinoid receptors and limited inhibitory effects on pyruvate dehydrogenase kinases (PDKs). It has been reported to possess a strong lysosomotropic property; this feature enables its assembly inside the acidic compartments within a cell, such as lysosomes, which may eventually hinder endocytosis.
1. Introduction
2. Chemistry
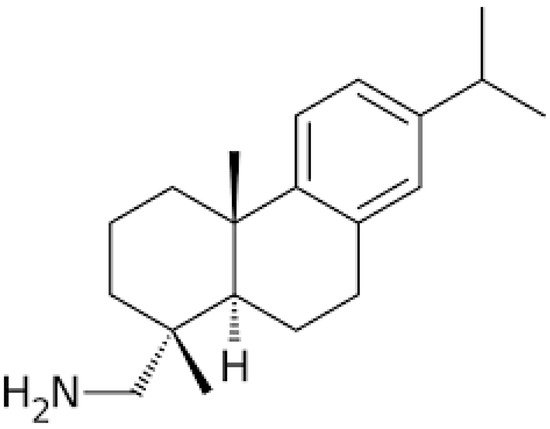
3. Anticancer Effects of Leelamine
References
- Jung, Y.Y.; Hwang, S.T.; Sethi, G.; Fan, L.; Arfuso, F.; Ahn, K.S. Potential Anti-Inflammatory and Anti-Cancer Properties of Farnesol. Molecules 2018, 23, 2827.
- Merarchi, M.; Sethi, G.; Fan, L.; Mishra, S.; Arfuso, F.; Ahn, K.S. Molecular Targets Modulated by Fangchinoline in Tumor Cells and Preclinical Models. Molecules 2018, 23, 2538.
- Sethi, G.; Shanmugam, M.K.; Warrier, S.; Merarchi, M.; Arfuso, F.; Kumar, A.P.; Bishayee, A. Pro-Apoptotic and Anti-Cancer Properties of Diosgenin: A Comprehensive and Critical Review. Nutrients 2018, 10, 645.
- Ko, J.H.; Sethi, G.; Um, J.Y.; Shanmugam, M.K.; Arfuso, F.; Kumar, A.P.; Bishayee, A.; Ahn, K.S. The Role of Resveratrol in Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 2589.
- Tewari, D.; Nabavi, S.F.; Nabavi, S.M.; Sureda, A.; Farooqi, A.A.; Atanasov, A.G.; Vacca, R.A.; Sethi, G.; Bishayee, A. Targeting activator protein 1 signaling pathway by bioactive natural agents: Possible therapeutic strategy for cancer prevention and intervention. Pharmacol. Res. 2018, 128, 366–375.
- Bishayee, A.; Sethi, G. Bioactive natural products in cancer prevention and therapy: Progress and promise. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2016; Volume 40–41, pp. 1–3.
- Shanmugam, M.K.; Lee, J.H.; Chai, E.Z.; Kanchi, M.M.; Kar, S.; Arfuso, F.; Dharmarajan, A.; Kumar, A.P.; Ramar, P.S.; Looi, C.Y.; et al. Cancer prevention and therapy through the modulation of transcription factors by bioactive natural compounds. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2016; Volume 40–41, pp. 35–47.
- Shanmugam, M.K.; Kannaiyan, R.; Sethi, G. Targeting cell signaling and apoptotic pathways by dietary agents: Role in the prevention and treatment of cancer. Nutr. Cancer 2011, 63, 161–173.
- Shrimali, D.; Shanmugam, M.K.; Kumar, A.P.; Zhang, J.; Tan, B.K.; Ahn, K.S.; Sethi, G. Targeted abrogation of diverse signal transduction cascades by emodin for the treatment of inflammatory disorders and cancer. Cancer Lett. 2013, 341, 139–149.
- Merarchi, M.; Sethi, G.; Shanmugam, M.K.; Fan, L.; Arfuso, F.; Ahn, K.S. Role of Natural Products in Modulating Histone Deacetylases in Cancer. Molecules 2019, 24, 1047.
- Yang, M.H.; Jung, S.H.; Sethi, G.; Ahn, K.S. Pleiotropic Pharmacological Actions of Capsazepine, a Synthetic Analogue of Capsaicin, against Various Cancers and Inflammatory Diseases. Molecules 2019, 24, 995.
- Jung, Y.Y.; Shanmugam, M.K.; Narula, A.S.; Kim, C.; Lee, J.H.; Namjoshi, O.A.; Blough, B.E.; Sethi, G.; Ahn, K.S. Oxymatrine Attenuates Tumor Growth and Deactivates STAT5 Signaling in a Lung Cancer Xenograft Model. Cancers 2019, 11, 49.
- Shanmugam, M.K.; Ahn, K.S.; Hsu, A.; Woo, C.C.; Yuan, Y.; Tan, K.H.B.; Chinnathambi, A.; Alahmadi, T.A.; Alharbi, S.A.; Koh, A.P.F.; et al. Thymoquinone Inhibits Bone Metastasis of Breast Cancer Cells Through Abrogation of the CXCR4 Signaling Axis. Front. Pharmacol. 2018, 9, 1294.
- Gou, Q.; Gong, X.; Jin, J.; Shi, J.; Hou, Y. Peroxisome proliferator-activated receptors (PPARs) are potential drug targets for cancer therapy. Oncotarget 2017, 8, 60704–60709.
- Shanmugam, M.K.; Manu, K.A.; Ong, T.H.; Ramachandran, L.; Surana, R.; Bist, P.; Lim, L.H.; Kumar, A.P.; Hui, K.M.; Sethi, G. Inhibition of CXCR4/CXCL12 signaling axis by ursolic acid leads to suppression of metastasis in transgenic adenocarcinoma of mouse prostate model. Int. J. Cancer 2011, 129, 1552–1563.
- Liu, L.; Ahn, K.S.; Shanmugam, M.K.; Wang, H.; Shen, H.; Arfuso, F.; Chinnathambi, A.; Alharbi, S.A.; Chang, Y.; Sethi, G.; et al. Oleuropein induces apoptosis via abrogating NF-kappaB activation cascade in estrogen receptor-negative breast cancer cells. J. Cell. Biochem. 2019, 120, 4504–4513.
- Deorukhkar, A.; Krishnan, S.; Sethi, G.; Aggarwal, B.B. Back to basics: How natural products can provide the basis for new therapeutics. Expert Opin. Investig. Drugs 2007, 16, 1753–1773.
- Bhuvanalakshmi, G.; Rangappa, K.S.; Dharmarajan, A.; Sethi, G.; Kumar, A.P.; Warrier, S. Breast Cancer Stem-Like Cells Are Inhibited by Diosgenin, a Steroidal Saponin, by the Attenuation of the Wnt beta-Catenin Signaling via the Wnt Antagonist Secreted Frizzled Related Protein-4. Front. Pharmacol. 2017, 8, 124.
- Jia, L.Y.; Shanmugam, M.K.; Sethi, G.; Bishayee, A. Potential role of targeted therapies in the treatment of triple-negative breast cancer. Anti-Cancer Drugs 2016, 27, 147–155.
- Woo, C.C.; Hsu, A.; Kumar, A.P.; Sethi, G.; Tan, K.H. Thymoquinone inhibits tumor growth and induces apoptosis in a breast cancer xenograft mouse model: The role of p38 MAPK and ROS. PLoS ONE 2013, 8, e75356.
- Manu, K.A.; Shanmugam, M.K.; Rajendran, P.; Li, F.; Ramachandran, L.; Hay, H.S.; Kannaiyan, R.; Swamy, S.N.; Vali, S.; Kapoor, S.; et al. Plumbagin inhibits invasion and migration of breast and gastric cancer cells by downregulating the expression of chemokine receptor CXCR4. Mol. Cancer 2011, 10, 107.
- Lee, H.; Baek, S.H.; Lee, J.H.; Kim, C.; Ko, J.H.; Lee, S.G.; Chinnathambi, A.; Alharbi, S.A.; Yang, W.M.; Um, J.Y.; et al. Isorhynchophylline, a Potent Plant Alkaloid, Induces Apoptotic and Anti-Metastatic Effects in Human Hepatocellular Carcinoma Cells through the Modulation of Diverse Cell Signaling Cascades. Int. J. Mol. Sci. 2017, 18, 1095.
- Ko, J.H.; Um, J.Y.; Lee, S.G.; Yang, W.M.; Sethi, G.; Ahn, K.S. Conditioned media from adipocytes promote proliferation, migration, and invasion in melanoma and colorectal cancer cells. J. Cell. Physiol. 2019, 234, 18249–18261.
- Singh, S.S.; Yap, W.N.; Arfuso, F.; Kar, S.; Wang, C.; Cai, W.; Dharmarajan, A.M.; Sethi, G.; Kumar, A.P. Targeting the PI3K/Akt signaling pathway in gastric carcinoma: A reality for personalized medicine? World J. Gastroenterol. 2015, 21, 12261–12273.
- Lee, J.H.; Kim, C.; Lee, S.G.; Yang, W.M.; Um, J.Y.; Sethi, G.; Ahn, K.S. Ophiopogonin D modulates multiple oncogenic signaling pathways, leading to suppression of proliferation and chemosensitization of human lung cancer cells. Phytomed. Int. J. Phytother. Phytopharm. 2018, 40, 165–175.
- Wang, N.; Pan, W.; Zhu, M.; Zhang, M.; Hao, X.; Liang, G.; Feng, Y. Fangchinoline induces autophagic cell death via p53/sestrin2/AMPK signalling in human hepatocellular carcinoma cells. Br. J. Pharmacol. 2011, 164, 731–742.
- Lu, S.; Sung, T.; Lin, N.; Abraham, R.T.; Jessen, B.A. Lysosomal adaptation: How cells respond to lysosomotropic compounds. PLoS ONE 2017, 12, e0173771.
- Kuzu, O.F.; Gowda, R.; Sharma, A.; Robertson, G.P. Leelamine mediates cancer cell death through inhibition of intracellular cholesterol transport. Mol. Cancer Ther. 2014, 13, 1690–1703.
- Hillhouse, T.M.; Porter, J.H. A brief history of the development of antidepressant drugs: From monoamines to glutamate. Exp. Clin. Psychopharmacol. 2015, 23, 1–21.
- Kocsis, J.H.; Frances, A.J.; Voss, C.; Mann, J.J.; Mason, B.J.; Sweeney, J. Imipramine treatment for chronic depression. Arch. Gen. Psychiatry 1988, 45, 253–257.
- Rainsford, K.D.; Parke, A.L.; Clifford-Rashotte, M.; Kean, W.F. Therapy and pharmacological properties of hydroxychloroquine and chloroquine in treatment of systemic lupus erythematosus, rheumatoid arthritis and related diseases. Inflammopharmacology 2015, 23, 231–269.
- Sharma, O.P. Effectiveness of chloroquine and hydroxychloroquine in treating selected patients with sarcoidosis with neurological involvement. Arch. Neurol. 1998, 55, 1248–1254.
- Lichtenfels, M.; Dornelles, A.D.S.; Petry, F.D.S.; Blank, M.; de Farias, C.B.; Roesler, R.; Schwartsmann, G. The anticancer estrogen receptor antagonist tamoxifen impairs consolidation of inhibitory avoidance memory through estrogen receptor alpha. J. Neural Transm. 2017, 124, 1331–1339.
- Brufsky, A.M.; Dickler, M.N. Estrogen Receptor-Positive Breast Cancer: Exploiting Signaling Pathways Implicated in Endocrine Resistance. Oncologist 2018, 23, 528–539.
- Cesen, M.H.; Repnik, U.; Turk, V.; Turk, B. Siramesine triggers cell death through destabilisation of mitochondria, but not lysosomes. Cell Death Dis. 2013, 4, e818.
- Armstrong, D.K.; Spriggs, D.; Levin, J.; Poulin, R.; Lane, S. Hematologic safety and tolerability of topotecan in recurrent ovarian cancer and small cell lung cancer: An integrated analysis. Oncologist 2005, 10, 686–694.
- HYCAMTIN® (Topotecan Hydrochloride) for Injection [Prescribing Information]; GlaxoSmithKline: Research Triangle Park, NC, USA, 2006.
- Rothenberg, M.L. Topoisomerase I inhibitors: Review and update. Ann. Oncol. 1997, 8, 837–855.
- von Pawel, J.; Schiller, J.H.; Shepherd, F.A.; Fields, S.Z.; Kleisbauer, J.P.; Chrysson, N.G.; Stewart, D.J.; Clark, P.I.; Palmer, M.C.; Depierre, A.; et al. Topotecan versus cyclophosphamide, doxorubicin, and vincristine for the treatment of recurrent small-cell lung cancer. J. Clin. Oncol. 1999, 17, 658–667.
- Garst, J. Topotecan: An evolving option in the treatment of relapsed small cell lung cancer. Ther. Clin. Risk Manag. 2007, 3, 1087–1095.
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal accumulation of anticancer drugs triggers lysosomal exocytosis. Oncotarget 2017, 8, 45117–45132.
- Le Tourneau, C.; Raymond, E.; Faivre, S. Sunitinib: A novel tyrosine kinase inhibitor. A brief review of its therapeutic potential in the treatment of renal carcinoma and gastrointestinal stromal tumors (GIST). Ther. Clin. Risk Manag. 2007, 3, 341–348.
- Gowda, R.; Inamdar, G.S.; Kuzu, O.; Dinavahi, S.S.; Krzeminski, J.; Battu, M.B.; Voleti, S.R.; Amin, S.; Robertson, G.P. Identifying the structure-activity relationship of leelamine necessary for inhibiting intracellular cholesterol transport. Oncotarget 2017, 8, 28260–28277.
- Liu, L.Y.; Alexa, K.; Cortes, M.; Schatzman-Bone, S.; Kim, A.J.; Mukhopadhyay, B.; Cinar, R.; Kunos, G.; North, T.E.; Goessling, W. Cannabinoid receptor signaling regulates liver development and metabolism. Development 2016, 143, 609–622.
- Lieberman, A.P.; Puertollano, R.; Raben, N.; Slaugenhaupt, S.; Walkley, S.U.; Ballabio, A. Autophagy in lysosomal storage disorders. Autophagy 2012, 8, 719–730.
- Futerman, A.H.; van Meer, G. The cell biology of lysosomal storage disorders. Nat. Rev. Mol. Cell Biol. 2004, 5, 554–565.
- Sehrawat, A.; Kim, S.H.; Hahm, E.R.; Arlotti, J.A.; Eiseman, J.; Shiva, S.S.; Rigatti, L.H.; Singh, S.V. Cancer-selective death of human breast cancer cells by leelamine is mediated by bax and bak activation. Mol. Carcinog. 2017, 56, 337–348.
- Gowda, R.; Madhunapantula, S.V.; Kuzu, O.F.; Sharma, A.; Robertson, G.P. Targeting multiple key signaling pathways in melanoma using leelamine. Mol. Cancer Ther. 2014, 13, 1679–1689.
- Chen, Y.C.; Gowda, R.; Newswanger, R.K.; Leibich, P.; Fell, B.; Rosenberg, G.; Robertson, G.P. Targeting cholesterol transport in circulating melanoma cells to inhibit metastasis. Pigment Cell Melanoma Res. 2017, 30, 541–552.
- Singh, K.B.; Ji, X.; Singh, S.V. Therapeutic Potential of Leelamine, a Novel Inhibitor of Androgen Receptor and Castration-Resistant Prostate Cancer. Mol. Cancer Ther. 2018, 17, 2079–2090.
- Sim, J.; Nam, W.; Lee, D.; Lee, S.; Hungchan, O.; Joo, J.; Liu, K.H.; Han, J.Y.; Ki, S.H.; Jeong, T.C.; et al. Selective induction of hepatic cytochrome P450 2B activity by leelamine in vivo, as a potent novel inducer. Arch. Pharm. Res. 2015, 38, 725–733.
- Fedorenko, I.V.; Gibney, G.T.; Sondak, V.K.; Smalley, K.S. Beyond BRAF: Where next for melanoma therapy? Br. J. Cancer 2015, 112, 217–226.
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954.
- Gandhi, J.; Zhang, J.; Xie, Y.; Soh, J.; Shigematsu, H.; Zhang, W.; Yamamoto, H.; Peyton, M.; Girard, L.; Lockwood, W.W.; et al. Alterations in genes of the EGFR signaling pathway and their relationship to EGFR tyrosine kinase inhibitor sensitivity in lung cancer cell lines. PLoS ONE 2009, 4, e4576.
- Chan, M.M.; Haydu, L.E.; Menzies, A.M.; Azer, M.W.; Klein, O.; Lyle, M.; Clements, A.; Guminski, A.; Kefford, R.F.; Long, G.V. The nature and management of metastatic melanoma after progression on BRAF inhibitors: Effects of extended BRAF inhibition. Cancer 2014, 120, 3142–3153.
- McCain, J. The MAPK (ERK) Pathway: Investigational Combinations for the Treatment Of BRAF-Mutated Metastatic Melanoma. Pharm. Ther. Peer Rev. J. Formul. Manag. 2013, 38, 96–108.
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Murray, T.; Thun, M.J. Cancer statistics, 2008. CA Cancer J. Clin. 2008, 58, 71–96.
- Hotte, S.J.; Saad, F. Current management of castrate-resistant prostate cancer. Curr. Oncol. 2010, 17 (Suppl. 2), S72–S79.
- Lee, J.H.; Kim, C.; Baek, S.H.; Ko, J.H.; Lee, S.G.; Yang, W.M.; Um, J.Y.; Sethi, G.; Ahn, K.S. Capsazepine inhibits JAK/STAT3 signaling, tumor growth, and cell survival in prostate cancer. Oncotarget 2017, 8, 17700–17711.
- Zhang, J.; Ahn, K.S.; Kim, C.; Shanmugam, M.K.; Siveen, K.S.; Arfuso, F.; Samym, R.P.; Deivasigamanim, A.; Lim, L.H.; Wang, L.; et al. Nimbolide-Induced Oxidative Stress Abrogates STAT3 Signaling Cascade and Inhibits Tumor Growth in Transgenic Adenocarcinoma of Mouse Prostate Model. Antioxid. Redox Signal. 2016, 24, 575–589.
- Mostaghel, E.A.; Page, S.T.; Lin, D.W.; Fazli, L.; Coleman, I.M.; True, L.D.; Knudsen, B.; Hess, D.L.; Nelson, C.C.; Matsumoto, A.M.; et al. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: Therapeutic implications for castration-resistant prostate cancer. Cancer Res. 2007, 67, 5033–5041.


