For the last couple of decades, natural products, either applied singly or in conjunction with other cancer therapies including chemotherapy and radiotherapy, have allowed us to combat different types of human cancers through the inhibition of their initiation and progression. The principal sources of these useful compounds are isolated from plants that were described in traditional medicines for their curative potential. Leelamine, derived from the bark of pine trees, was previously reported as having a weak agonistic effect on cannabinoid receptors and limited inhibitory effects on pyruvate dehydrogenase kinases (PDKs). It has been reported to possess a strong lysosomotropic property; this feature enables its assembly inside the acidic compartments within a cell, such as lysosomes, which may eventually hinder endocytosis.
1. Introduction
Cancer remains a major cause of mortality on a global scale. Moreover, among the patients who respond positively to the treatment, most develop recurrence after a certain delay, leading to severe complications during therapy. Various bioactive natural products can exert significant beneficial effects in cancer treatment and prevention [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15,
16]. Indeed, natural products constitute an important source for the discovery of anticancer compounds, as they can modulate various important hallmarks of tumor cells, and several antineoplastic drugs approved by the United States’ Food and Drug Administration (FDA) over the last four decades are based primarily on natural products and/or their derivatives [
4,
7,
8,
13,
14,
17,
18,
19,
20,
21,
22,
23,
24,
25,
26].
The process of lysosomotropism can be defined as the accumulation of compounds with basic and lipophilic properties inside the lysosomes. Lysosomes are acidic single-membrane-enclosed organelles filled with peptidases including cathepsins that are necessary for the digestion of macromolecules, including diverse xenobiotics, via different degradation pathways such as endocytosis, phagocytosis, and so on. Lysosomes are responsible for cellular homoeostasis, and recent studies have also shown their implication in nutrient sensing and regulation of cellular metabolism and proliferation. Lysosomotropism can lead to the disturbance of homeostasis and disorder in the lysosomes and endosomes that impacts various lysosome-dependent cellular processes, including autophagy and cholesterol trafficking and thus growth and proliferation, among others [
27,
28]. Interestingly, numerous lysosomotropic compounds have shown therapeutic properties and are mainly used in psychiatry as antidepressants and antipsychotics (e.g., chloropromazine, approved in 1999 by the FDA [
29], and imipramine and Tofranil®, approved in 1959 by the FDA). Imipramine is a tricyclic compound derived from dibenzazepine; this antidepressant enables the inhibition of two neurotransmitters’ reuptake (serotonin and norepinephrine) within the synaptic cleft of the central nervous system (CNS) [
30]. Moreover, imipramine is suggested not only for depression therapy, but can also be employed for childhood nighttime incontinence, a condition that can cause psychological distress in children [
30]. Interestingly, lysosomotropic products may be used as antimalarial compounds (chloroquine phosphate, ARALEN®, approved in 1949 by the FDA). Interestingly, chloroquine was applied to treat malaria and other inflammation-associated disorders initially [
31], but the discovery of the autophagy process led to its potential application in cancer.
Thereafter, significant lysosomal damage was highlighted in animals receiving chloroquine, and thus it was classified as a lysosomotropic. It could also readily penetrate the lysosomal membrane, thus causing its accumulation within the lysosome [
32]. Other lysosomotropic agents such as estrogen receptor antagonists (tamoxifen and Nolvadex®, approved in 2002 by the FDA), may exhibit both estrogenic agonistic and antagonistic effects in different parts of the body. Due to these antagonistic properties, it can be used as a selective estrogen receptor modulator. In the breast tissue, it acts as an estrogen antagonist and causes antiestrogenic and anti-breast cancer effects. Through downstream intracellular processes, it can modulate processes occurring during the cell cycle, which also leads to its classification as a cytostatic compound. In the bone, tamoxifen can stimulate estrogen receptors instead of blocking them, thus exerting an estrogen agonistic effect, and may prevent osteoporosis in postmenopausal women [
33,
34].
Interestingly, recent studies have also demonstrated that tumor cells were sensitive to some lysosomotropic products, and this property has been described as a promising approach for the selective inhibition of tumor cells. Among such compounds, siramesine has shown notable anticancer effects with nonsignificant toxicity targeting diverse tumor cell lines [
35]. Siramesine, at concentrations above 20 µM, has been shown to induce a loss of matrix metallopeptidases (MMPs), apparently by a direct destabilization of the mitochondrial membrane, and afterwards by triggering cell death, both apoptotic and nonapoptotic. Interestingly, at concentrations below 20 µM, a slower cell death occurred, linked to the gradual deterioration of cellular homeostasis due to interference in diverse activities, such as lysosomal degradation, autophagic flux, intracellular trafficking, and energy metabolism [
35].
Topotecan, a topoisomerase inhibitor can be applied for the treatment of metastatic ovarian cancer as well as that of platinum-sensitive relapsed small cell lung cancer [
36]. This agent was also recently approved for therapy of stage IVB recurrent or persistent cervical cancer [
37]. Topotecan can attach to topoisomerase I and form a complex with DNA, thus causing breaks during DNA replication and therby promote cell death [
38]. Moreover, topotecan has also exhibited symptom palliation in small cell lung cancer (SCLC) patients, leading to an improvement in dyspnea, anorexia, hoarseness, fatigue, and interference with daily activities [
39,
40]. Sunitinib malate is a multitargeted tyrosine kinase inhibitor that can display significant anticancer effects. It has also been tested for its possible lysosomotropic property that may also confer significant anticancer activity, and has shown promising results in human cervical cancer cells [
41,
42].
2. Chemistry
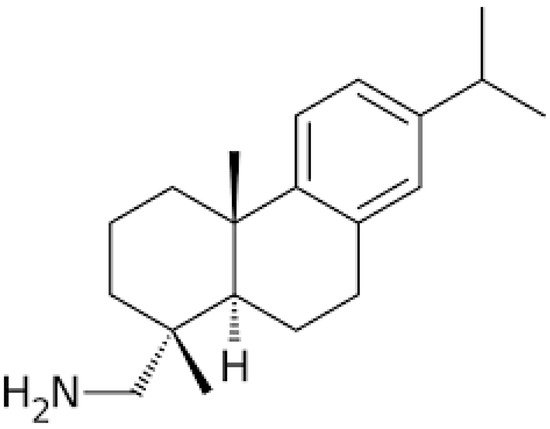
Among the identified lysosomotropic compounds, leelamine (dehydroabietylamine), a lipophilic diterpene amine phytochemical with a pKa of 9.9, is a natural compound extracted from the bark of pine trees [
28,
43]. Leelamine (structure shown in
Figure 1), which was previously reported as having a weak agonistic effect on cannabinoid receptors and limited inhibitory effects on pyruvate dehydrogenase kinases (PDKs) [
28,
44], has caught particular attention for its lysosomotropic property and significant inhibitory effects on proliferation and tumorigenesis [
43,
45,
46] in human breast cancer cells [
47], melanoma cell lines [
28,
43,
48,
49], and prostate cancer cell lines [
50]. A recent study, during which chemical modifications of leelamine were made to identify its active site responsible for its anticancer activities, has shown that a suppression of the amino group or its charge may lead to a deletion of its antineoplastic actions, thus indicating that its lysosomotropic as well as its antitumorigenic effect may be mediated by this moiety [
28]. Furthermore, a recent discovery made by Sehrawat et al. has also suggested a novel potential application of leelamine as a potent antidiabetic agent through the induction of cytochrome P450 2B6 (CYP2B) activity [
51].
Figure 1. The chemical structure of leelamine.
3. Anticancer Effects of Leelamine
Malignant melanoma has the ability to undergo rapid metastasis and also an aptitude for developing chemotherapeutic resistance [
52]. V600E-BRAF is a mutant of the B Rapidly Accelerated Fibrosarcoma protein with a aline residue replaced by glutamic acid in the kinase domain, which leads to an upregulation of the kinase activity and drug resistance development. Among the used treatments, zelboraf, tafinlar, and mekinist can target the deregulated MAPK pathway [
53,
54]. Nevertheless, those therapeutic strategies have seen their efficiency limited by drug resistance that causes the evolution of further aggressive melanomas [
55,
56].
In melanoma cancer cell lines treated with leelamine, an inhibition of tumor development without significant systemic toxicity has been shown using various techniques, including modulation transfer spectroscopy MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] assay and liquid chromatography–mass spectrometry [
43]; furthermore, a decrease in the proliferation and vascular development has been highlighted [
48]. Nanolipolee-007, a liposomal form of leelamine, was also shown to reduce melanoma metastasis and to induce apoptosis [
49]. Prostate cancer represents 25% of all cancers diagnosed in men and its incidence continues to rise in many countries; it is also the fifth most frequent cause of mortality among all the cancer types in men [
57,
58,
59,
60]. Castration-resistant prostate cancer (CRPC) resulting from androgen withdrawal has been found to be quite challenging to cure due to its resistance to testosterone-suppression therapy, which is supposed to be the most efficient castration treatment [
61].
Indeed, CRPC can be driven by aberrant activation of the androgen receptor (AR) by mechanisms varying from its amplification, mutation, and post-translational modification to expression of splice variants, including AR-V7. Leelamine treatment of prostate cancer cell lines LNCaP (an androgen-responsive cell line), C4-2B (an androgen-insensitive variant of LNCaP), and 22Rv1 (a CRPC cell line that express AR-Vs), and a murine prostate cancer cell line, Myc-CaP, led to decreased mitotic activity and prostate-specific antigen expression, in addition to apoptosis induction that caused cell death [
50]. Breast cancer is major first cause of cancer mortality among women at a global scale, and even though the effectiveness of the existing multidisciplinary approaches to treat breast cancer has been proved, its therapy still remains a great medical challenge [
18,
19,
20,
21]. In MDA-MB-231, MCF-7, and SUM159 breast cancer cells, it has been shown that leelamine provoked cancer cell death through an apoptotic process in a dose-dependent fashion, while a normal mammary epithelial cell line (MCF-10A) was found to be completely resistant to the apoptotic and cell growth-inhibiting effects of the drug. Interestingly, it has been demonstrated that leelamine was also able to suppress breast cancer stem cells’ self-renewal ability substantially [
47].
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines7030053