 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Alice Ranjan | + 4508 word(s) | 4508 | 2021-06-26 07:31:16 | | | |
| 2 | Alice Ranjan | -112 word(s) | 4396 | 2021-07-01 22:11:09 | | | | |
| 3 | Conner Chen | Meta information modification | 4396 | 2021-07-02 07:27:02 | | |
Video Upload Options
Glioblastoma is the most common and aggressive primary malignant brain tumor, and more than two-thirds of patients with glioblastoma die within two years of diagnosis. The challenges of treating this disease mainly include genetic and microenvironmental features that often render the tumor resistant to treatments. Despite extensive research efforts, only a small number of drugs tested in clinical trials have become therapies for patients. Targeting cyclin-dependent kinase 9 (CDK9) is an emerging therapeutic approach that has the potential to overcome the challenges in glioblastoma management.
1. CDK9: An Important Regulator of Transcription Elongation
CDK9 is broadly expressed in all types of human tissues and is present in two isoforms in mammalian cells: CDK9-49 and CDK9-55, which differ only by their molecular weight, but functionally are both able to associate with cyclins T1, T2A, T2B, or K (with CDK9 binding primarily to cyclin T1) [1]. The CDK9-cyclin T1 complex forms the positive transcription elongation factor b (P-TEFb), which plays a crucial role in regulating transcription elongation (Figure 1) [2]. Shortly after the initiation of transcription, RNA Pol II pauses at the promoter-proximal region, located 30–60 nucleotides downstream of the transcription start site [3]. This pausing of RNA Pol II serves as a quality control step to allow for 5′-capping and other modifications and is facilitated by promoter-associated transcription factors, negative elongation factor (NELF), and DRB-sensitivity-inducing factor (DSIF). For elongation to continue and for mature mRNA to be generated, the paused RNA Pol II must be released from the promoter-proximal site, and P-TEFb serves as a main regulator of this step. In order for P-TEFb to be fully activated, CDK9 is first phosphorylated by CDK7 at Threonine 186 [4], and subsequently, P-TEFb phosphorylates Serine 2 of RNA Pol II’s carboxyl-terminal domain (CTD), NELF, DSIF, and the CTD-linker of RNA Pol II in order to release RNA Pol II [5][6].
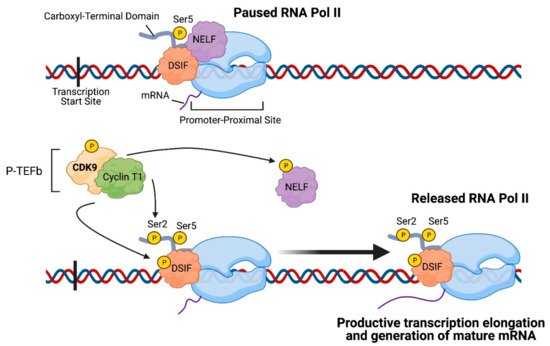
Figure 1. Role of CDK9 in transcription elongation: Positive transcription elongation factor b (P-TEFb), which is composed of cyclin-dependent kinase 9 (CDK9) and Cyclin T1, phosphorylates Serine 2 on the carboxyl-terminal domain of RNA Polymerase II (RNA Pol II) as well as negative elongation factor (NELF) and DRB-sensitivity-inducing factor (DSIF). Consequently, RNA Polymerase II is released from the promoter-proximal site and engages in productive transcription elongation and generation of mature mRNA. The image was created with BioRender.com (accessed on 18 April 2021).
P-TEFb can exist in two other states in the cell—either reversibly bound in an inhibitory complex consisting of HEXIM1/2 and the small nuclear ribonucleoprotein (snRNP) 7SK or assembled with other transcription factors in an active super elongation complex (SEC) that interacts with RNA Pol II’s CTD [3]. The mechanism by which P-TEFb is released from the inhibitory complex and recruited to the promoter-proximal site is mediated by Jumonji Domain Containing 6 (JMJD6) and Bromodomain-containing protein 4 (BRD4), with JMJD6 binding directly with CDK9 and BRD4 with Cyclin T1 [5]. Notably, an shRNA loss-of-function screen demonstrated that glioblastoma cells in an orthotopic xenograft mouse model, but not in vitro, were dependent on JMJD6 and BRD4 for survival [7]. Furthermore, expression of JMJD6 was shown to increase with glioma grade and inhibiting JMJD6 extended survival of the glioma-bearing mice [7]. Additionally, a study demonstrated that two multi-CDK inhibitors, flavopiridol, and SNS-032, reduced levels of phosphorylated RNA Pol II in glioblastoma cells and disrupted anchorage-independent growth and cancer cell migration, two hallmarks of cancer cells [8][9]. These results reveal that glioblastomas are dependent on RNA Pol II pause-release and CDK9-containing complexes for their survival and present a rationale for further exploration of targeting CDK9 and its interacting factors as a therapeutic approach.
2. Impact of CDK9 Inhibition on Cancer Cells
In this section, we review studies in glioblastoma and other cancer types that demonstrate how CDK9 inhibition can modulate various cancer cell survival pathways to facilitate an anti-tumor response. Importantly, CDK9 inhibition does not specifically target different subsets of genes in different tumors. Rather, many of the pathways that are impacted by CDK9 inhibition in the various cancers reflect the different pre-established gene expression profiles of their tissues of origin.
2.1. Transcription
Given that CDK9 plays a crucial role in regulating transcription elongation, inhibiting CDK9 can reduce the transcription of genes necessary for maintaining cancer cell survival (Figure 2A). Su et al. demonstrated that zotiraciclib (a multi-kinase inhibitor that primarily targets CDK9) suppressed phosphorylation of CDK9 and RNA Pol II in glioblastoma cells, which resulted in decreased transcription of anti-apoptotic proteins such as MCL-1 and Survivin (encoded by BIRC5) and induced activation of caspase-3, resulting in cell apoptosis [4]. Furthermore, overexpression of a constitutively active CDK9 mutant rescued glioblastoma cells from zotiraciclib-induced cell death [4]. Le Rhun et al. note, however, that caspase activation is not essential for zotiraciclib-induced cell death [10]. The authors observed that glioblastoma cells co-exposed to zotiraciclib and a pan-caspase inhibitor demonstrated only moderately weakened zotiraciclib-induced cytotoxicity, and zotiraciclib still suppressed phosphorylation of RNA Pol II and depleted MCL-1 protein levels [10]. While the authors found that zotiraciclib-induced cell death occurs in a caspase-independent manner, they suggest that caspase inhibition may be involved in delaying cell death and that the proximate cause of death may be through zotiraciclib-induced metabolic alterations in glioblastoma cells, as demonstrated by Su et al. [4][10].
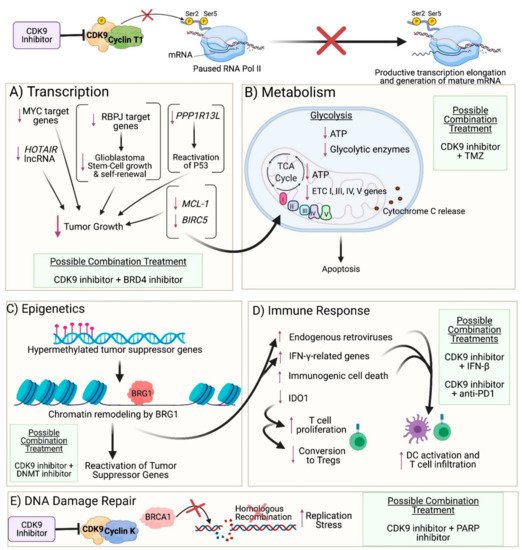
Figure 2. Impact of CDK9 inhibition on cancer cells: (A) Transcription: CDK9 inhibition prevents phosphorylation of Serine 2 on the carboxyl-terminal domain of RNA Polymerase II (RNA Pol II), thereby preventing productive transcription elongation of genes that are critical to the survival and proliferation of cancer cells, such as MYC target genes, HOTAIR lncRNA, PPP1R13L, RBPJ target genes, MCL-1, and BIRC5. (B) Metabolism: Reduced levels of anti-apoptotic proteins encoded by MCL-1 and BIRC5 result in mitochondrial dysfunction and damage as observed by the downregulated expression of genes involved in respiratory complexes I, III, IV, and V and the release of cytochrome c into the cytoplasm. Furthermore, CDK9 inhibition leads to reduced expression of glycolytic enzymes and ATP levels generated from both glycolysis and oxidative phosphorylation, ultimately leading to apoptosis. (C) Epigenetics: CDK9 inhibition enables recruitment of BRG1 to heterochromatin, where BRG1 remodels nucleosomes in order to facilitate transcription of genes, resulting in reactivation of tumor suppressor genes that were previously silenced by hypermethylation in their promoter region. (D) Immune Response: BRG1-mediated chromatin remodeling also results in reactivation of endogenous retroviruses and expression of IFN-γ-related genes. Moreover, CDK9 inhibition results in immunogenic tumor cell death and decreased expression of IDO1. Collectively, these events result in activation of dendritic cells (DCs) and proliferation and recruitment of effector T cells while reducing the conversion of T cells into regulatory T cells. (E) DNA Damage Repair: Inhibition of the CDK9-Cyclin K complex (but not the CDK9-Cyclin T1 complex) prevents CDK9-dependent BRCA1 recruitment to double-strand DNA breaks, thus precluding homologous recombination and resulting in increased replication stress. (A–E): Possible combination treatments involving CDK9 inhibitors are listed. The image was created with BioRender.com (accessed on 18 April 2021).
CDK9 inhibition has also been reported as a useful strategy for treating cancers with MYC overexpression. MYC is of particular interest as a target in glioblastoma, given that it regulates about 15% of the entire genome, modulating cell proliferation, differentiation, survival, and apoptosis [11], and dysregulation in MYC signaling contributes to tumorigenesis [12]. However, MYC also maintains the proliferation of non-tumor cells and lacks effective binding pockets for small-molecule drugs, making its direct pharmacological inhibition challenging [11]. This underscores the need for methods that can indirectly inhibit MYC signaling, such as potentially targeting transcription of the MYC gene instead [11]. Importantly, transcription elongation of MYC is largely dependent on P-TEFb-mediated promoter-pause release [13]. Furthermore, MYC recruits P-TEFb to promoters to enhance transcription of its target genes, and MYC-overexpressing tumor cells are dependent on this activity [13]. A clinical trial for the highly selective CDK9 inhibitor KB-0742 was even launched recently in January 2021 to treat MYC-amplified cancers and is currently enrolling patients with advanced solid tumors or non-Hodgkin’s lymphoma [14]. Preclinically, for glioblastomas, zotiraciclib has been shown in one study to potently suppress the growth of MYC-overexpressing glioblastoma cells, and higher MYC expression was correlated with greater sensitivity to the drug [12]. However, another study demonstrated no correlation between gene silencing of MYC and sensitivity to zotiraciclib in glioblastoma cells [10]. Furthermore, zotiraciclib was shown to have varying effects on MYC transcript and protein expression levels: mRNA and protein expression decreased in some cell lines but increased in others following treatment and also varied based on the concentration of drug administered and time of exposure to the drug [10]. It is possible that CDK9 inhibitors may interfere more with MYC’s activity as a transcription factor than with transcription of the MYC gene. Furthermore, given that many pathways can activate MYC expression, it is possible that MYC expression can increase even with CDK9 inhibition due to other factors that possibly bypass promoter-proximal pausing, such as Aurora kinase A, which functions as a transactivating factor through its interaction with heterogeneous nuclear ribonucleoprotein K to activate MYC expression [15]. A study in HeLa cells similarly demonstrated an increase in MYC expression following treatment with i-CDK9, an inhibitor selective for CDK9, and the authors proposed that the increase in MYC expression may be part of a cellular compensatory mechanism to cope with CDK9 inhibition and to ensure maximal expression of important genes that are controlled by both MYC and CDK9 [16]. Moreover, BRD4 may be key to facilitating this compensatory mechanism –not only is BRD4 important in recruiting P-TEFb from the inhibitory 7SK snRNP/HEXIM1/2 complex to the promoter-proximal site, but it was shown in the study to use its C-terminal P-TEFb-interaction domain (PID) to directly increase CDK9′s catalytic activity and to render CDK9 more resistant to inhibition [16]. Furthermore, BRD4 inhibition was shown to reduce the interaction between BRD4 and CDK9 at the MYC locus and prevented the increase in MYC expression caused by i-CDK9 in HeLa, lung cancer, and melanoma cells [16]. Importantly, inhibition of both CDK9 and BRD4 exhibited a synergistic effect through the induction of apoptosis in HeLa and non-small cell lung cancer cells [16]. These findings thus suggest the value of targeting both CDK9 and BRD4 in MYC-overexpressing cells.
CDK9 inhibition has important implications not only for the oncogene MYC but for the tumor suppressor P53 as well. Loss of P53 function frequently occurs in the development of cancer either through mutations in the TP53 gene or inhibition of the wild-type p53 protein by negative regulators [17]. The inhibitor of apoptosis-stimulating protein of p53 (iASPP), encoded by PPP1R13L, is one such negative regulator. Higher expression of the iASPP-SV isoform has been found to correlate with malignancy in gliomas, and glioblastomas may increase expression of iASPP-SV in order to promote tumor progression and prevent apoptosis [18]. Notably, glioma patients with high iASPP-SV expression experience lower overall survival and 5-year progression-free survival than patients with low iASPP-SV expression [18]. In a study conducted in a colon cancer cell line, the CDK9 inhibitors SNS-032, flavopiridol, and LDC067 were shown to downregulate transcription of iASPP, resulting in reactivation of the tumor-suppressive function of wild-type p53 [17]. The findings from this study may provide a promising CDK9-based strategy to counteract the pro-tumor effects of iASPP-SV in glioblastomas as well.
CDK9-mediated phosphorylation of RNA Pol II has also been demonstrated to be involved in a positive feedback loop that contributes to the upregulation and persistent expression of the long non-coding RNA HOX Transcript Antisense RNA (HOTAIR), which is overexpressed in multiple cancers and contributes to cancer progression [19]. HOTAIR has also been shown to promote malignant progression in gliomas and serves as a negative prognostic factor for survival of glioma patients [20]. A study demonstrated that pancreatic ductal adenocarcinoma, hepatocellular carcinoma, and colorectal cancer cells were unable to produce full-length, functional HOTAIR transcripts following CDK9 inhibition by LDC067 [19]. Targeting CDK9 may thus provide a mechanism to reduce HOTAIR expression in glioblastomas as well. Furthermore, it has been shown that BRD4 binds to the HOTAIR promoter and that treatment of glioblastoma cells with I-BET151 (an inhibitor against BRD4 and others in the bromodomain and extraterminal domain (BET) protein family) reduced levels of HOTAIR transcripts [21]. This provides yet another reason for the combined use of CDK9 and BRD4 inhibitors to synergistically inhibit proliferation of glioblastoma cells.
Additionally, CDK9 inhibition may provide a therapeutic approach to inhibit transcription of genes involved in maintaining GSCs. Efforts to target GSCs have often focused on inhibiting NOTCH since NOTCH signaling is critical in determining stem cell fate and cancer [22]. However, phase I trials investigating NOTCH antagonists in gliomas have shown limited efficacy [23]. An alternative solution may involve the recombination signal binding protein for immunoglobulin kappa J region (RBPJ), a transcription effector in NOTCH signaling that regulates a distinct transcription program compared to NOTCH [23]. Importantly, it was shown in a study that RBPJ relies on CDK9-mediated transcription elongation and that CDK9 inhibition results in decreased GSC growth and self-renewal [23]. Since GSCs are critical players in tumor formation and resistance to treatment, this finding supports the utility of targeting CDK9 to reduce treatment resistance caused by GSCs.
2.2. Metabolism
Targeting CDK9 has also been shown to induce metabolic stress in glioblastomas (Figure 2B). As previously discussed, zotiraciclib decreased transcription of the anti-apoptotic proteins MCL-1 and Survivin in glioblastoma cells, resulting in apoptosis [4]. Importantly, MCL-1 and Survivin also play vital roles in maintaining the function and integrity of the mitochondria. Zotiraciclib was shown to induce mitochondrial dysfunction in glioblastoma cells, as observed by the downregulation in expression of most genes involved in respiratory complexes I, III, IV, and V [4]. Moreover, zotiraciclib altered mitochondrial membrane potential and disrupted mitochondrial membrane integrity, as observed by the presence of dysmorphic mitochondria under electron microscopy and the release of cytochrome c into the cytoplasm by both Raman imaging and western blot [4]. Notably, zotiraciclib-induced mitochondrial damage was potentiated when TMZ treatment was included as well [4]. Zotiraciclib was also shown to suppress glycolysis, resulting in depleted intracellular ATP levels, and combination treatment of zotiraciclib and TMZ demonstrated a synergistic effect through further glycolytic suppression, as observed by the downregulation of the glycolytic enzymes Hexokinase 2 (HK2), Pyruvate Kinase isoform M2 (PKM2), and Lactate Dehydrogenase A (LDHA), which are usually highly expressed in glioblastomas [4][24][25][26]. Since HK2 and PKM2 promote tumor growth and GSC self-renewal [24][25] and glioblastomas are heavily reliant on glycolysis for energy production [27], targeting CDK9 provides a novel mechanism to exploit these metabolic vulnerabilities. Furthermore, since targeting CDK9 resulted in mitochondrial damage, glioblastoma cells are limited in their ability to compensate for energy production via oxidative phosphorylation. Inhibition of both energy production pathways thus increases the likelihood of cancer cell death.
2.3. DNA Damage Repair
Although CDK9 is often associated with transcription elongation, it also plays an important role in pathways that maintain genomic integrity [2]. Furthermore, Cyclin K, but not Cyclin T, is involved with CDK9 in these DNA damage repair processes: CDK9-Cyclin K interacts with DNA damage repair proteins such as ataxia telangiectasia and Rad3-related protein and accumulates on chromatin to limit the generation of single-stranded DNA resulting from DNA damage [2]. The CDK9-55 isoform, but not CDK9-42, has also been shown to associate with Ku70, a protein involved in double-strand DNA break (DSB) repair, and shRNA depletion of CDK9-55 in HeLa cells resulted in apoptosis and DSBs [28].
Given the importance of CDK9 and Cyclin K in DNA damage repair, studies in cancer models have focused on inhibiting CDK9 to generate increased replication stress and facilitate cancer cell death. In one study, osteosarcoma cells pre-treated with either the CDK9 inhibitor flavopiridol or 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole demonstrated impaired cell cycle recovery following treatment with hydroxyurea [29]. Another study conducted in head and neck squamous cell carcinoma (HNSCC) cell lines examined the relationship between CDK9, DNA damage, and sensitivity to radiation [30]. siRNA knockdown of CDK9 resulted in significant induction of γH2AX (indicating DSBs), delayed cell cycle transition, and increased sensitivity to radiation. In contrast, overexpression of CDK9 enhanced the survival of HNSCC cells that were treated with radiation [30].
Glioblastomas demonstrate high levels of DNA replication, which predisposes them to significant replication stress [31]. BRCA1 is usually considered a tumor suppressor, but in glioblastomas, BRCA1 helps to mitigate replication stress and extend cancer cell survival [31]. High expression of BRCA1 has been shown to correlate with lower overall survival in glioblastoma patients [31], underscoring the need to investigate treatments that can dysregulate BRCA1 signaling. Importantly, CDK9 has been shown to modulate the recruitment of BRCA1 to DSBs and plays an important role in the BRCA1-mediated homologous recombination (HR) DNA repair process (Figure 2E) [32]. Cell survival assays performed in wild-type BRCA1 mammary gland carcinoma cells, for example, demonstrated that knockdown of CDK9 impaired recruitment of BRCA1 to DSBs and sensitized cells to radiation [32].
Tumors with mutations in BRCA1 are deficient in HR and are often vulnerable to treatment with inhibitors of Poly(ADP-ribose) polymerase (PARP), another enzyme involved in DNA repair processes [33]. Research has focused on expanding the benefit of PARP inhibitors to wild-type BRCA1 cancer cells by combining PARP inhibitors with other treatment options in order to achieve a synthetic lethal effect [33]. In one study, wild-type BRCA1 mammary gland carcinoma cells were treated with an shRNA targeting CDK9 and were found to be more sensitive to the combined treatment of radiation and Olaparib, a PARP inhibitor, than cells with intact CDK9 [32]. Another study in wild-type BRCA1 ovarian cancer cells demonstrated that the combined treatment of Olaparib and the CDK9 inhibitor CDKI-73 suppressed colony formation and induced apoptosis, and additionally reduced tumor growth in a xenograft mouse model [34]. Importantly, CDKI-73 was shown to downregulate BRCA1 expression, which contributed to increased sensitivity to Olaparib [34]. These studies present important implications for the treatment of glioblastomas: The majority of glioblastomas are reported to carry wild-type BRCA1 and are proficient in homologous recombination, and consequently, PARP inhibitors have shown limited efficacy [35]. Since BRCA1 is dependent on CDK9 for HR repair, CDK9 inhibition may provide a synthetic lethal mechanism to render glioblastomas more vulnerable to PARP inhibitors.
2.4. Epigenetics
Tumor suppressor genes (TSGs) in cancers are silenced via epigenetic modifications. Specifically, methylation in TSG promoters recruits repressor complexes that ultimately lead to the formation of heterochromatin [36]. Zhang et al. demonstrated that CDK9 maintains gene silencing in cancer cells by phosphorylating BRG1, a component of the SWI/SNF chromatin remodeling complex, preventing BRG1 from being recruited to the heterochromatin to move and restructure nucleosomes and mediate gene transcription [36]. Importantly, CDK9 inhibition was shown to dephosphorylate BRG1, enabling BRG1 to access and remodel the chromatin, leading to the reactivation of TSGs (Figure 2C) [36]. The authors also examined the anti-tumor effects of CDK9 inhibition in an ovarian cancer mouse model treated with SNS-032 and observed reactivation of ovarian-cancer specific hypermethylated TSGs, decreased tumor burden, and prolonged survival [36].
Glioblastomas exhibit frequent hypermethylation of TSGs, such as RB1, EMP3, RASSF1A, and BLU [37]. Moreover, one study reported that significant hypermethylation of the pro-apoptotic CASP8 occurred during the progression from primary to recurrent glioblastoma, which possibly conferred a growth advantage to tumor cells remaining after radiation/chemotherapy treatment [38]. Targeting CDK9 may thus provide a mechanism to reactivate TSGs in glioblastomas and to counteract hypermethylation of factors that could render the tumor resistant to future treatments. Interestingly, Zhang et al. also demonstrated that the transcriptional profile induced by CDK9 inhibition was similar to the transcriptional profile induced by treatment with a DNA methyltransferase inhibitor (DNMTi), which demethylated the promoters of silenced TSGs, allowing for reactivation of these genes [36]. Combining CDK9 inhibition with DNMT inhibition may thus attain synergistic induction of TSGs and inhibition of tumor growth.
2.5. Immune Response
Given the immunosuppressive tumor microenvironment (TME) of glioblastomas, there is a need to identify better combination therapies that can improve the immune response against the tumor. Interferon (IFN)-β plays an immunosuppressive role in the TME by countering the pro-inflammatory effects of IFN-γ and preventing T cell trafficking into the CNS [39]. Interestingly, one study found that pre-exposure to IFN-β rendered glioblastoma cells more sensitive to subsequent treatment with zotiraciclib [40]. Specifically, combined treatment of IFN-β and zotiraciclib increased inhibition of cell growth compared to zotiraciclib treatment alone. The combined treatment also suppressed phosphorylation of RNA Pol II and reduced protein expression of CDK9 to a greater extent than zotiraciclib did alone, though how IFN-β mediates this synergistic effect remains to be explored [40]. On a separate but related note, a pharmacokinetic study of zotiraciclib, conducted as part of a phase I trial for patients with recurrent anaplastic astrocytoma and glioblastoma (NCT02942264), revealed a significant increase in patient plasma concentrations of cytokines, including IP-10, at 24 h after an oral dose of zotiraciclib [41]. IP-10 has been reported to promote an anti-tumor immune response by attracting cytotoxic T and NK cells [39], though whether this occurs following zotiraciclib treatment remains to be determined. The induction of IP-10, along with the synergistic effect of IFN-β and zotiraciclib on glioblastoma cells, indicates the potential of zotiraciclib to counteract the immunosuppressive TME as one of the mechanisms of its anti-glioma effects.
The role of CDK9 inhibition in modulating the immune system (Figure 2D) has been further elucidated by Zhang et al. In one of their studies, colon cancer cells were treated with the CDK9 inhibitor HH1, and RNA sequencing of the cells identified upregulation of 326 immune-related genes [36]. Among these were endogenous retroviruses (ERVs), genetic elements originating from retroviruses that infected our ancestral germline and that are rarely expressed in healthy cells but can become expressed in cancer cells due to epigenetic dysregulation [42]. Importantly, the expression of ERVs may mimic a viral infection and induce IFN, thereby serving as potential tumor-associated antigens that can activate cytotoxic T cells [42]. Indeed, the 326-immune related genes from Zhang et al.’s study included genes in the IFN-γ pathway as well as the Major Histocompatibility Complex genes/Human Leukocyte Antigens HLA-A, HLA-B, and HLA-C [36]. Regarding glioblastoma, ERVs may serve as potential biomarkers for treatment given that a study characterizing the profile of ERVs in glioblastoma found 46 differentially expressed ERVs between glioblastoma and normal brain tissue, with 43 of those ERVs upregulated in glioblastoma [43].
Zhang et al. further demonstrated in an ovarian cancer mouse model that CDK9 inhibition via SNS-032 led to an increase in CD45+ immune cells, CD3+ T cells, and activated dendritic cells in the TME [36]. Furthermore, CDK9 inhibition sensitized the cells to anti-PD1 treatment, as demonstrated by an increased immune response following combination treatment [36].
Zhang et al. also observed in the TCGA database that colon cancer and melanoma patients with high expression of the same immune genes that were upregulated following CDK9 inhibition demonstrated significantly longer survival than patients with low expression of these genes [36]. This survival data, along with the prior results, provides evidence that targeting CDK9 may counteract the immunosuppressive TME and sensitize tumors to immune checkpoint inhibitors to yield potential clinical benefit.
CDK9 inhibition has also been shown to enhance the immune response against tumors by inducing immunogenic cell death (ICD) in tumor cells [44]. ICD occurs when dying cells release or express on their surface certain danger-associated molecular patterns that can activate an immune response [44]. Classic features of ICD include translocation of calreticulin (which is normally restricted to the endoplasmic reticulum (ER)) to the cell surface, the extracellular release of high mobility group box1 (HMGB1) and ATP, and activation of the Type I IFN pathway [44][45]. Altogether, ICD has the potential to induce dendritic cell (DC) activation and, ultimately, cross-presentation of antigens to cytotoxic T cells [45]. The multi-CDK inhibitor dinaciclib suppresses transcription of MCL-1 (which typically protects cells from ER stress) and was shown to elicit ICD in colon adenocarcinoma cells, as observed by the cell-surface expression of calreticulin and release of HMGB1 and ATP [44]. Furthermore, dinaciclib stimulated transient expression of Type I IFN response genes when administered as a single-agent treatment and in combination with anti-PD1 [44]. Importantly, combination treatment of dinaciclib and anti-PD1 resulted in increased DC activation and T cell infiltration and inhibition of tumor growth in a colon adenocarcinoma mouse model compared to dinaciclib and anti-PD1 single-agent treatments [44].
In addition to its ability to synergize with anti-PD1, dinaciclib can downregulate the expression of the immunosuppressive enzyme indoleamine 2,3-dioxygenase (IDO) in glioblastoma cells, as shown in a study by Riess et al. [46]. IDO is produced in response to IFN-γ and plays an important role in tryptophan metabolism by mediating the degradation of tryptophan and accumulation of kynurenine [47]. IDO-activity has been shown to inhibit T cell proliferation, promote T cell apoptosis, and induce regulatory T cells [47]. Glioblastomas exhibit high expression of IDO1, and increased IDO1 expression serves as a negative prognostic factor for patient survival [47]. Interestingly, Riess et al. also demonstrated that IDO1 expression increased in two glioblastoma cell lines following TMZ treatment and that IDO1 expression was reduced in TMZ-treated cells after additional treatment with dinaciclib [46]. This suggests the potential utility of combining CDK9 inhibitors with the standard treatment of TMZ in order to mitigate any immunosuppressive effects induced by TMZ via IDO1.
It is interesting to note that CDK9 inhibition can upregulate the expression of IFN-γ-stimulated genes [36] yet also suppress the activity of IDO1, an IFN-γ-stimulated gene [46]. CDK9 inhibition may serve then as a strategy to upregulate IFN-γ-stimulated genes that promote anti-tumor effects while mitigating immunosuppressive effects elicited by IDO1.
While the above studies highlight the advantages of inhibiting CDK9 in order to mediate a pro-inflammatory response against cancer cells, systemic blockade of CDK9 activity also has the potential to suppress the adaptive immune system. In fact, CDK9 inhibition has been investigated as an anti-inflammatory therapeutic approach for inflammatory conditions, such as arthritis [48], since CDK9 inhibition was shown to increase the percentage of regulatory T cells in spleens from arthritic mice [48]. Furthermore, flavopiridol inhibited the recruitment of NF-κB (a pro-inflammatory transcription factor that binds to P-TEFb to stimulate transcription elongation) in human endothelial cells [48]. This led to reduced ICAM-1 expression, which is important for recruiting lymphocytes to sites of inflammation [48]. In another study, zotiraciclib was shown to abrogate B cell receptor (BCR) signaling, though this provides a strategy for treating chronic lymphocytic leukemia (CLL) since leukemia cell survival is partly sustained through constitutive activation of BCR signaling [49]. Furthermore, CDK9 inhibition has the potential to adversely affect T cell activation. The dual CDC7/CDK9 inhibitor PHA-767491/NMS-1116354 was shown to affect signal transduction downstream of the T cell receptor (TCR) by inhibiting Erk phosphorylation, which is important for T cell activation and degrading the p105 isoform of NF-κB, which is important for regulating T cell homeostasis [50]. The inhibitory effects of CDK9 on T cell signaling and function have been speculated to contribute to the adverse immune-related symptoms that patients receiving CDK inhibitors in clinical trials may experience [50]. Further studies focusing on the optimal timing and dosage that allows for an immunogenic response against tumor cells without detrimental effects on T cell proliferation and effector functions are warranted.
References
- Morales, F.; Giordano, A. Overview of CDK9 as a target in cancer research. Cell Cycle 2016, 15, 519–527.
- Yu, D.S.; Cortez, D. A role for cdk9-cyclin k in maintaining genome integrity. Cell Cycle 2011, 10, 28–32.
- Jonkers, I.; Lis, J.T. Getting up to speed with transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2015, 16, 167–177.
- Su, Y.-T.; Chen, R.; Wang, H.; Song, H.; Zhang, Q.; Chen, L.-Y.; Lappin, H.; Vasconcelos, G.; Lita, A.; Maric, D.; et al. Novel Targeting of Transcription and Metabolism in Glioblastoma. Clin. Cancer Res. 2018, 24, 1124.
- Liu, W.; Ma, Q.; Wong, K.; Li, W.; Ohgi, K.; Zhang, J.; Aggarwal, A.; Rosenfeld, M.G. Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell 2013, 155, 1581–1595.
- Vos, S.M.; Farnung, L.; Boehning, M.; Wigge, C.; Linden, A.; Urlaub, H.; Cramer, P. Structure of activated transcription complex Pol II–DSIF–PAF–SPT6. Nature 2018, 560, 607–612.
- Miller, T.E.; Liau, B.B.; Wallace, L.C.; Morton, A.R.; Xie, Q.; Dixit, D.; Factor, D.C.; Kim, L.J.Y.; Morrow, J.J.; Wu, Q.; et al. Transcription elongation factors represent in vivo cancer dependencies in glioblastoma. Nature 2017, 547, 355–359.
- Bhutada, I.; Chellappan, S.; Padmanabhan, J. Abstract 2310: Targeting transcription-associated CDKs is an effective way to combat glioblastoma and medulloblastoma with minimal effect on primary neurons. Cancer Res. 2018, 78, 2310.
- Guadamillas, M.C.; Cerezo, A.; del Pozo, M.A. Overcoming anoikis—Pathways to anchorage-independent growth in cancer. J. Cell Sci. 2011, 124, 3189.
- Le Rhun, E.; von Achenbach, C.; Lohmann, B.; Silginer, M.; Schneider, H.; Meetze, K.; Szabo, E.; Weller, M. Profound, durable and MGMT-independent sensitivity of glioblastoma cells to cyclin-dependent kinase inhibition. Int. J. Cancer 2019, 145, 242–253.
- Wang, C.; Zhang, J.; Yin, J.; Gan, Y.; Xu, S.; Gu, Y.; Huang, W. Alternative approaches to target Myc for cancer treatment. Signal Transduct. Target. Ther. 2021, 6, 117.
- Tsang, J.; Sung, S.; Gosa, L.; Meetze, K.; Cloughesy, T.; Nathanson, D. EXTH-67. TG02, A brain-penetrant multi-cdk inhibitor, potently suppresses myc-driven glioblastoma. Neuro-Oncology 2017, 19, vi87–vi88.
- Huang, C.-H.; Lujambio, A.; Zuber, J.; Tschaharganeh, D.F.; Doran, M.G.; Evans, M.J.; Kitzing, T.; Zhu, N.; de Stanchina, E.; Sawyers, C.L.; et al. CDK9-mediated transcription elongation is required for MYC addiction in hepatocellular carcinoma. Genes Dev. 2014, 28, 1800–1814.
- A Dose Escalation and Cohort Expansion Study of KB-0742 in Participants with Relapsed or Refractory Solid Tumors or Non-Hodgkin Lymphoma. Available online: (accessed on 15 April 2021).
- Zheng, F.; Yue, C.; Li, G.; He, B.; Cheng, W.; Wang, X.; Yan, M.; Long, Z.; Qiu, W.; Yuan, Z.; et al. Nuclear AURKA acquires kinase-independent transactivating function to enhance breast cancer stem cell phenotype. Nat. Commun. 2016, 7, 10180.
- Lu, H.; Xue, Y.; Yu, G.K.; Arias, C.; Lin, J.; Fong, S.; Faure, M.; Weisburd, B.; Ji, X.; Mercier, A.; et al. Compensatory induction of MYC expression by sustained CDK9 inhibition via a BRD4-dependent mechanism. eLife 2015, 4, e06535.
- Wu, J.; Liang, Y.; Tan, Y.; Tang, Y.; Song, H.; Wang, Z.; Li, Y.; Lu, M. CDK9 inhibitors reactivate p53 by downregulating iASPP. Cell Signal. 2020, 67, 109508.
- Liu, X.; Kang, J.; Liu, F.; Wen, S.; Zeng, X.; Liu, K.; Luo, Y.; Ji, X.; Zhao, S. Overexpression of iASPP-SV in glioma is associated with poor prognosis by promoting cell viability and antagonizing apoptosis. Tumor Biol. 2016, 37, 6323–6330.
- Wong, C.H.; Li, C.H.; He, Q.; Tong, J.H.M.; To, K.-F.; Chen, Y. The Establishment of CDK9/ RNA PolII/H3K4me3/DNA Methylation Feedback Promotes HOTAIR Expression by RNA Elongation Enhancement in Cancer. bioRxiv 2019, 812776.
- Zhou, X.; Ren, Y.; Zhang, J.; Zhang, C.; Zhang, K.; Han, L.; Kong, L.; Wei, J.; Chen, L.; Yang, J.; et al. HOTAIR is a therapeutic target in glioblastoma. Oncotarget 2015, 6, 8353–8365.
- Pastori, C.; Kapranov, P.; Penas, C.; Peschansky, V.; Volmar, C.-H.; Sarkaria, J.N.; Bregy, A.; Komotar, R.; St. Laurent, G.; Ayad, N.G.; et al. The Bromodomain protein BRD4 controls HOTAIR, a long noncoding RNA essential for glioblastoma proliferation. Proc. Natl. Acad. Sci. USA 2015, 112, 8326.
- Wang, J.; Wakeman, T.P.; Lathia, J.D.; Hjelmeland, A.B.; Wang, X.-F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch Promotes Radioresistance of Glioma Stem Cells. Stem Cells 2010, 28, 17–28.
- Xie, Q.; Wu, Q.; Kim, L.; Miller, T.E.; Liau, B.B.; Mack, S.C.; Yang, K.; Factor, D.C.; Fang, X.; Huang, Z.; et al. RBPJ maintains brain tumor–initiating cells through CDK9-mediated transcriptional elongation. J. Clin. Investig. 2016, 126, 2757–2772.
- Wolf, A.; Agnihotri, S.; Munoz, D.; Guha, A. Developmental profile and regulation of the glycolytic enzyme hexokinase 2 in normal brain and glioblastoma multiforme. Neurobiol. Dis. 2011, 44, 84–91.
- Yang, W.; Xia, Y.; Ji, H.; Zheng, Y.; Liang, J.; Huang, W.; Gao, X.; Aldape, K.; Lu, Z. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature 2011, 480, 118–122.
- Di, H.; Zhang, X.; Guo, Y.; Shi, Y.; Fang, C.; Yuan, Y.; Wang, J.; Shang, C.; Guo, W.; Li, C. Silencing LDHA inhibits proliferation, induces apoptosis and increases chemosensitivity to temozolomide in glioma cells. Oncol. Lett. 2018, 15, 5131–5136.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674.
- Liu, H.; Herrmann, C.H.; Chiang, K.; Sung, T.-L.; Moon, S.-H.; Donehower, L.A.; Rice, A.P. 55K isoform of CDK9 associates with Ku70 and is involved in DNA repair. Biochem. Biophys. Res. Commun. 2010, 397, 245–250.
- Yu, D.S.; Zhao, R.; Hsu, E.L.; Cayer, J.; Ye, F.; Guo, Y.; Shyr, Y.; Cortez, D. Cyclin-dependent kinase 9–cyclin K functions in the replication stress response. Embo. Rep. 2010, 11, 876–882.
- Storch, K.; Cordes, N. The impact of CDK9 on radiosensitivity, DNA damage repair and cell cycling of HNSCC cancer cells. Int. J. Oncol. 2016, 48, 191–198.
- Rasmussen, R.D.; Gajjar, M.K.; Tuckova, L.; Jensen, K.E.; Maya-Mendoza, A.; Holst, C.B.; Møllgaard, K.; Rasmussen, J.S.; Brennum, J.; Bartek, J.; et al. BRCA1-regulated RRM2 expression protects glioblastoma cells from endogenous replication stress and promotes tumorigenicity. Nat. Commun. 2016, 7, 13398.
- Nepomuceno, T.C.; Fernandes, V.C.; Gomes, T.T.; Carvalho, R.S.; Suarez-Kurtz, G.; Monteiro, A.N.; Carvalho, M.A. BRCA1 recruitment to damaged DNA sites is dependent on CDK9. Cell Cycle 2017, 16, 665–672.
- Ning, J.-F.; Stanciu, M.; Humphrey, M.R.; Gorham, J.; Wakimoto, H.; Nishihara, R.; Lees, J.; Zou, L.; Martuza, R.L.; Wakimoto, H.; et al. Myc targeted CDK18 promotes ATR and homologous recombination to mediate PARP inhibitor resistance in glioblastoma. Nat. Commun. 2019, 10, 2910.
- Li, J.; Zhi, X.; Chen, S.; Shen, X.; Chen, C.; Yuan, L.; Guo, J.; Meng, D.; Chen, M.; Yao, L. CDK9 inhibitor CDKI-73 is synergetic lethal with PARP inhibitor olaparib in BRCA1 wide-type ovarian cancer. Am. J. Cancer Res. 2020, 10, 1140–1155.
- Sizemore, S.T.; Mohammad, R.; Sizemore, G.M.; Nowsheen, S.; Yu, H.; Ostrowski, M.C.; Chakravarti, A.; Xia, F. Synthetic Lethality of PARP Inhibition and Ionizing Radiation is p53-dependent. Mol. Cancer Res. 2018, 16, 1092.
- Zhang, H.; Pandey, S.; Travers, M.; Sun, H.; Morton, G.; Madzo, J.; Chung, W.; Khowsathit, J.; Perez-Leal, O.; Barrero, C.A.; et al. Targeting CDK9 Reactivates Epigenetically Silenced Genes in Cancer. Cell 2018, 175, 1244–1258.e1226.
- Carén, H.; Pollard, S.M.; Beck, S. The good, the bad and the ugly: Epigenetic mechanisms in glioblastoma. Mol. Asp. Med. 2013, 34, 849–862.
- Martinez, R.; Schackert, G. Epigenetic Aberrations in Malignant Gliomas: An Open Door Leading to Better Understanding and Treatment. Epigenetics 2007, 2, 147–150.
- Ratnam, N.M.; Gilbert, M.R.; Giles, A.J. Immunotherapy in CNS cancers: The role of immune cell trafficking. Neuro-Oncology 2018, 21, 37–46.
- Lohmann, B.; Le Rhun, E.; Silginer, M.; Epskamp, M.; Weller, M. Interferon-β sensitizes human glioblastoma cells to the cyclin-dependent kinase inhibitor, TG02. Oncol. Lett. 2020, 19, 2649–2656.
- Wu, J.; Yuan, Y.; Long Priel, D.A.; Fink, D.; Peer, C.J.; Sissung, T.M.; Su, Y.-T.; Pang, Y.; Yu, G.; Butler, M.K.; et al. Phase I Study of Zotiraciclib in Combination with Temozolomide for Patients with Recurrent High-grade Astrocytomas. Clin. Cancer Res. 2021.
- Attermann, A.S.; Bjerregaard, A.M.; Saini, S.K.; Grønbæk, K.; Hadrup, S.R. Human endogenous retroviruses and their implication for immunotherapeutics of cancer. Ann. Oncol. 2018, 29, 2183–2191.
- Yuan, Z.; Zhang, N.; An, Z.; Zheng, W. Abstract B37: Analysis of the differential expression of human endogenous retrovirus in glioblastoma multiforme. Cancer Res. 2020, 80, B37.
- Hossain, D.M.S.; Javaid, S.; Cai, M.; Zhang, C.; Sawant, A.; Hinton, M.; Sathe, M.; Grein, J.; Blumenschein, W.; Pinheiro, E.M.; et al. Dinaciclib induces immunogenic cell death and enhances anti-PD1-mediated tumor suppression. J. Clin. Investig. 2018, 128, 644–654.
- Rajani, K.R.; Carlstrom, L.P.; Parney, I.F.; Johnson, A.J.; Warrington, A.E.; Burns, T.C. Harnessing Radiation Biology to Augment Immunotherapy for Glioblastoma. Front Oncol. 2019, 8, 656.
- Riess, C.; Schneider, B.; Kehnscherper, H.; Gesche, J.; Irmscher, N.; Shokraie, F.; Classen, C.F.; Wirthgen, E.; Domanska, G.; Zimpfer, A.; et al. Activation of the Kynurenine Pathway in Human Malignancies Can Be Suppressed by the Cyclin-Dependent Kinase Inhibitor Dinaciclib. Front. Immunol. 2020, 11.
- Woroniecka, K.I.; Rhodin, K.E.; Chongsathidkiet, P.; Keith, K.A.; Fecci, P.E. T-cell Dysfunction in Glioblastoma: Applying a New Framework. Clin. Cancer Res. 2018, 24, 3792.
- Hellvard, A.; Zeitlmann, L.; Heiser, U.; Kehlen, A.; Niestroj, A.; Demuth, H.-U.; Koziel, J.; Delaleu, N.; Jan, P.; Mydel, P. Inhibition of CDK9 as a therapeutic strategy for inflammatory arthritis. Sci. Rep. 2016, 6, 31441.
- Chen, R.; Tsai, J.; Thompson, P.A.; Chen, Y.; Xiong, P.; Liu, C.; Burrows, F.; Sivina, M.; Burger, J.A.; Keating, M.J.; et al. The multi-kinase inhibitor TG02 induces apoptosis and blocks B-cell receptor signaling in chronic lymphocytic leukemia through dual mechanisms of action. Blood Cancer J. 2021, 11, 57.
- Chen, E.W.; Tay, N.Q.; Brzostek, J.; Gascoigne, N.R.J.; Rybakin, V. A Dual Inhibitor of Cdc7/Cdk9 Potently Suppresses T Cell Activation. Front. Immunol. 2019, 10.

