 +1 credit
+1 credit
Video Upload Options
Despite the tremendous efforts made in the past decades, severe side/toxic effects and poor bioavailability still represent the main challenges that hinder the clinical translation of drug molecules. This has turned the attention of investigators towards drug delivery vehicles that provide a localized and controlled drug delivery. Molecularly imprinted polymers (MIPs) as novel and versatile drug delivery vehicles have been widely studied in recent years due to the advantages of selective recognition, enhanced drug loading, sustained release, and robustness in harsh conditions. This review highlights the design and development of strategies undertaken for MIPs used as drug delivery vehicles involving different drug delivery mechanisms, such as rate-programmed, stimuli-responsive and active targeting, published during the course of the past five years.
1. Introduction
1.1. Drug Delivery Systems
1.2. Molecular Imprinting: Advanced Synthetic Molecular Recognition
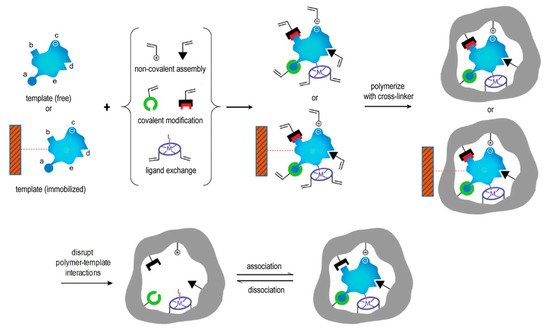
1.3. The Rationale of MIPs Used in Drug Delivery
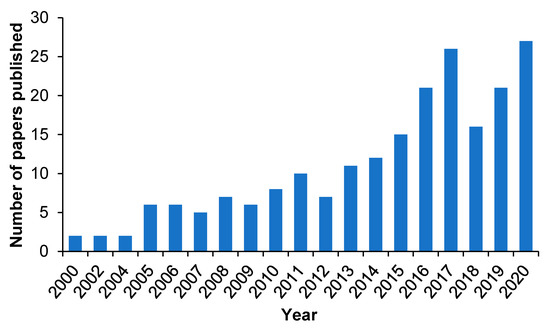
2. MIP-Based Drug Delivery Systems
2.1. Rate-Programmed Drug Delivery
2.2. Stimuli-Responsive Drug Delivery
2.3. Active Targeting Drug Delivery
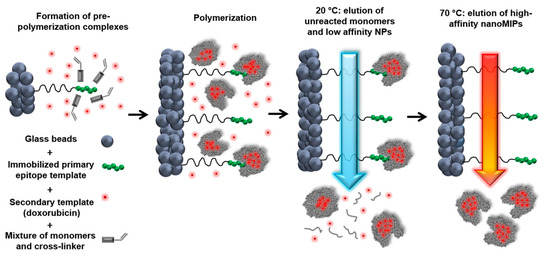
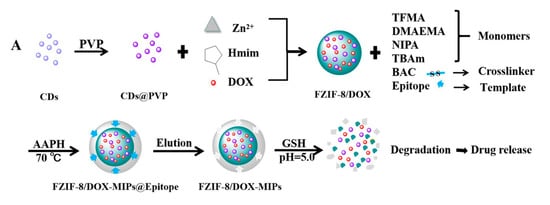
3. Current Challenges in MIP-Based DDS
4. Conclusions and Future Perspectives
References
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12.
- Wen, H.; Jung, H.; Li, X. Drug Delivery Approaches in Addressing Clinical Pharmacology-Related Issues: Opportunities and Challenges. AAPS J. 2015, 17, 1327–1340.
- Alvarez-Lorenzo, C.; Concheiro, A. Molecularly imprinted polymers for drug delivery. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2004, 804, 231–245.
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem. Rev. 2016, 116, 2602–2663.
- Zaidi, S.A. Molecular imprinted polymers as drug delivery vehicles. Drug Deliv. 2016, 23, 2262–2271.
- Oz, U.C.; Bolat, Z.B.; Poma, A.; Guan, L.; Telci, D.; Sahin, F.; Battaglia, G.; Bozkır, A. Prostate cancer cell-specific BikDDA delivery by targeted polymersomes. Appl. Nanosci. 2020, 10, 3389–3401.
- Kim, E.-S.; Kim, D.; Nyberg, S.; Poma, A.; Cecchin, D.; Jain, S.A.; Kim, K.-A.; Shin, Y.-J.; Kim, E.-H.; Kim, M.; et al. LRP-1 functionalized polymersomes enhance the efficacy of carnosine in experimental stroke. Sci. Rep. 2020, 10, 699.
- Fenaroli, F.; Robertson, J.D.; Scarpa, E.; Gouveia, V.M.; Di Guglielmo, C.; De Pace, C.; Elks, P.M.; Poma, A.; Evangelopoulos, D.; Canseco, J.O.; et al. Polymersomes Eradicating Intracellular Bacteria. ACS Nano 2020, 14, 8287–8298.
- Zhu, Y.; Poma, A.; Rizzello, L.; Gouveia, V.M.; Ruiz-Perez, L.; Battaglia, G.; Williams, C.K. Metabolically Active, Fully Hydrolysable Polymersomes. Angew. Chem. Int. Ed. Engl. 2019, 58, 4581–4586.
- Rodríguez-Arco, L.; Poma, A.; Ruiz-Pérez, L.; Scarpa, E.; Ngamkham, K.; Battaglia, G. Molecular bionics–engineering biomaterials at the molecular level using biological principles. Biomaterials 2019, 192, 26–50.
- Gouveia, V.M.; Rizzello, L.; Nunes, C.; Poma, A.; Ruiz-Perez, L.; Oliveira, A.; Reis, S.; Battaglia, G. Macrophage Targeting pH Responsive Polymersomes for Glucocorticoid Therapy. Pharmaceutics 2019, 11, 614.
- Poma, A.; Pei, Y.; Ruiz-Perez, L.; Rizzello, L.; Battaglia, G. Polymersomes: Synthesis and Applications. In Encyclopedia of Polymer Science and Technology; Wiley: Hoboken, NJ, USA, 2018; pp. 1–43.
- Ellis, E.; Zhang, K.; Lin, Q.; Ye, E.; Poma, A.; Battaglia, G.; Loh, X.J.; Lee, T.-C. Biocompatible pH-responsive nanoparticles with a core-anchored multilayer shell of triblock copolymers for enhanced cancer therapy. J. Mater. Chem. B 2017, 5, 4421–4425.
- Radenkovic, D.; Arjun, S.; Poma, A.; Nyberg, S.; Battaglia, B.; Yellon, D.M.; Davidson, S. 162 Polymersomes Functionalized with HSP70–Novel, Synthetic Cardioprotective Nanovesicles. Heart 2016, 102, A115.
- Scarpa, E.; De Pace, C.; Joseph, A.S.; de Souza, S.C.; Poma, A.; Liatsi-Douvitsa, E.; Contini, C.; De Matteis, V.; Martí, J.S.; Battaglia, G.; et al. Tuning cell behavior with nanoparticle shape. PLoS ONE 2020, 15, e0240197.
- Bueno, C.Z.; Apolinário, A.C.; Duro-Castano, A.; Poma, A.; Pessoa, A.; Rangel-Yagui, C.O.; Battaglia, G. l-Asparaginase Encapsulation into Asymmetric Permeable Polymersomes. ACS Macro Lett. 2020, 9, 1471–1477.
- Sood, N.; Bhardwaj, A.; Mehta, S.; Mehta, A. Stimuli-responsive hydrogels in drug delivery and tissue engineering. Drug Deliv. 2016, 23, 758–780.
- Vilar, G.; Tulla-Puche, J.; Albericio, F. Polymers and drug delivery systems. Curr. Drug Deliv. 2012, 9, 367–394.
- Mercadante, V.; Scarpa, E.; De Matteis, V.; Rizzello, L.; Poma, A. Engineering Polymeric Nanosystems against Oral Diseases. Molecules 2021, 26, 2229.
- Englert, C.; Brendel, J.C.; Majdanski, T.C.; Yildirim, T.; Schubert, S.; Gottschaldt, M.; Windhab, N.; Schubert, U.S. Pharmapolymers in the 21st century: Synthetic polymers in drug delivery applications. Prog. Polym. Sci. 2018, 87, 107–164.
- Liu, C.; Ewert, K.K.; Wang, N.; Li, Y.; Safinya, C.R.; Qiao, W. A multifunctional lipid that forms contrast-agent liposomes with dual-control release capabilities for precise MRI-guided drug delivery. Biomaterials 2019, 221, 119412.
- Cazzamalli, S.; Dal Corso, A.; Widmayer, F.; Neri, D. Chemically Defined Antibody- and Small Molecule-Drug Conjugates for in Vivo Tumor Targeting Applications: A Comparative Analysis. J. Am. Chem. Soc. 2018, 140, 1617–1621.
- Chen, T.; Ren, L.; Liu, X.; Zhou, M.; Li, L.; Xu, J.; Zhu, X. DNA Nanotechnology for Cancer Diagnosis and Therapy. Int. J. Mol. Sci. 2018, 19, 1671.
- Hoshino, Y.; Shea, K.J. The evolution of plastic antibodies. J. Mater. Chem. 2011, 21, 3517–3521.
- Lulinski, P. Molecularly imprinted polymers based drug delivery devices: A way to application in modern pharmacotherapy. A review. Mater. Sci. Eng. C 2017, 76, 1344–1353.
- Johan Svenson, I.A.N. On the thermal and chemical stability of molecularly imprinted polymers. Anal. Chim. Acta 2000, 435, 19–24.
- Saylan, Y.; Akgonullu, S.; Yavuz, H.; Unal, S.; Denizli, A. Molecularly Imprinted Polymer Based Sensors for Medical Applications. Sensors 2019, 19, 1279.
- Poma, A.; Turner, A.P.F.; Piletsky, S.A. Advances in the manufacture of MIP nanoparticles. Trends Biotechnol. 2010, 28, 629–637.
- Patel, K.D.; Kim, H.-W.; Knowles, J.C.; Poma, A. Molecularly Imprinted Polymers and Electrospinning: Manufacturing Convergence for Next-Level Applications. Adv. Funct. Mater. 2020, 30, 2001955.
- Polyakov, M. Adsorption properties and structure of silica gel. Zhur. Fiz. Khim. 1931, 2, 799–805.
- Wulff, G.; Sarhan, A. Über die Anwendung von enzymanalog gebauten Polymeren zur Racemattrennung. Angew. Chem. 1972, 84, 364.
- Arshady, R.; Mosbach, K. Synthesis of substrate-selective polymers by host-guest polymerization. Macromol. Chem. Phys. 1981, 182, 687–692.
- Wei, Y.; Zeng, Q.; Hu, Q.; Wang, M.; Tao, J.; Wang, L. Self-cleaned electrochemical protein imprinting biosensor basing on a thermo-responsive memory hydrogel. Biosens. Bioelectron. 2018, 99, 136–141.
- Malitesta, C.; Mazzotta, E.; Picca, R.A.; Poma, A.; Chianella, I.; Piletsky, S.A. MIP sensors—The electrochemical approach. Anal. Bioanal. Chem. 2012, 402, 1827–1846.
- Turner, N.W.; Bramhmbhatt, H.; Szabo-Vezse, M.; Poma, A.; Coker, R.; Piletsky, S.A. Analytical methods for determination of mycotoxins: An update (2009–2014). Anal. Chim. Acta 2015, 901, 12–33.
- Sellergren, B.; Allender, C.J. Molecularly imprinted polymers: A bridge to advanced drug delivery. Adv. Drug Deliv. Rev. 2005, 57, 1733–1741.
- Zhang, Z.; Liu, B.; Liu, J. Molecular Imprinting for Substrate Selectivity and Enhanced Activity of Enzyme Mimics. Small 2017, 13, 1602730.
- Nematollahzadeh, A.; Sun, W.; Aureliano, C.S.; Lutkemeyer, D.; Stute, J.; Abdekhodaie, M.J.; Shojaei, A.; Sellergren, B. High-capacity hierarchically imprinted polymer beads for protein recognition and capture. Angew. Chem. Int. Ed. 2011, 50, 495–498.
- Panagiotopoulou, M.; Kunath, S.; Medina-Rangel, P.X.; Haupt, K.; Tse Sum Bui, B. Fluorescent molecularly imprinted polymers as plastic antibodies for selective labeling and imaging of hyaluronan and sialic acid on fixed and living cells. Biosens. Bioelectron. 2017, 88, 85–93.
- Poma, A.; Whitcombe, M.; Piletsky, S. Plastic Antibodies. In Designing Receptors for the Next Generation of Biosensors; Piletsky, S.A., Whitcombe, M.J., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 105–129.
- Yu, Y.; Ye, L.; Haupt, K.; Mosbach, K. Formation of a class of enzyme inhibitors (drugs), including a chiral compound, by using imprinted polymers or biomolecules as molecular-scale reaction vessels. Angew. Chem. Int. Ed. 2002, 41, 4459–4463.
- Zaidi, S.A. Latest trends in molecular imprinted polymer based drug delivery systems. RSC Adv. 2016, 6, 88807–88819.
- Murakami, T.; Iwamuro, Y.; Ishimaru, R.; Chinaka, S.; Hasegawa, H. Molecularly imprinted polymer solid-phase extraction of synthetic cathinones from urine and whole blood samples. J. Sep. Sci. 2018, 41, 4506–4514.
- Ruela, A.L.M.; de Figueiredo, E.C.; Carvalho, F.C.; de Araújo, M.B.; Pereira, G.R. Adsorption and release of nicotine from imprinted particles synthesised by precipitation polymerisation: Optimising transdermal formulations. Eur. Polym. J. 2018, 100, 67–76.
- Bai, J.; Zhang, Y.; Chen, L.; Yan, H.; Zhang, C.; Liu, L.; Xu, X. Synthesis and characterization of paclitaxel-imprinted microparticles for controlled release of an anticancer drug. Mater. Sci. Eng. C 2018, 92, 338–348.
- Gagliardi, M.; Bertero, A.; Bifone, A. Molecularly Imprinted Biodegradable Nanoparticles. Sci. Rep. 2017, 7, 40046.
- Ceglowski, M.; Jerca, V.V.; Jerca, F.A.; Hoogenboom, R. Reduction-Responsive Molecularly Imprinted Poly(2-isopropenyl-2-oxazoline) for Controlled Release of Anticancer Agents. Pharmaceutics 2020, 12, 506.
- Piletsky, S.; Canfarotta, F.; Poma, A.; Bossi, A.M.; Piletsky, S. Molecularly Imprinted Polymers for Cell Recognition. Trends Biotechnol. 2020, 38, 368–387.
- Moczko, E.; Poma, A.; Guerreiro, A.; Perez de Vargas Sansalvador, I.; Caygill, S.; Canfarotta, F.; Whitcombe, M.J.; Piletsky, S. Surface-modified multifunctional MIP nanoparticles. Nanoscale 2013, 5, 3733–3741.
- Poma, A.; Guerreiro, A.; Whitcombe, M.J.; Piletska, E.V.; Turner, A.P.F.; Piletsky, S.A. Solid-Phase Synthesis of Molecularly Imprinted Polymer Nanoparticles with a Reusable Template–“Plastic Antibodies”. Adv. Funct. Mater. 2013, 23, 2821–2827.
- Subrahmanyam, S.; Guerreiro, A.; Poma, A.; Moczko, E.; Piletska, E.; Piletsky, S. Optimisation of experimental conditions for synthesis of high affinity MIP nanoparticles. Eur. Polym. J. 2013, 49, 100–105.
- Guerreiro, A.; Poma, A.; Karim, K.; Moczko, E.; Takarada, J.; de Vargas-Sansalvador, I.P.; Turner, N.; Piletska, E.; de Magalhães, C.S.; Glazova, N.; et al. Influence of surface-imprinted nanoparticles on trypsin activity. Adv. Healthc. Mater. 2014, 3, 1426–1429.
- Muzyka, K.; Karim, K.; Guerreiro, A.; Poma, A.; Piletsky, S. Optimisation of the synthesis of vancomycin-selective molecularly imprinted polymer nanoparticles using automatic photoreactor. Nanoscale Res. Lett. 2014, 9, 154.
- Poma, A.; Brahmbhatt, H.; Watts, J.K.; Turner, N.W. Nucleoside-Tailored Molecularly Imprinted Polymeric Nanoparticles (MIP NPs). Macromolecules 2014, 47, 6322–6330.
- Poma, A.; Guerreiro, A.; Caygill, S.; Moczko, E.; Piletsky, S. Automatic reactor for solid-phase synthesis of molecularly imprinted polymeric nanoparticles (MIP NPs) in water. RSC Adv. 2014, 4, 4203–4206.
- Poma, A.; Brahmbhatt, H.; Pendergraff, H.M.; Watts, J.K.; Turner, N.W. Generation of novel hybrid aptamer-molecularly imprinted polymeric nanoparticles. Adv. Mater. 2015, 27, 750–758.
- Canfarotta, F.; Poma, A.; Guerreiro, A.; Piletsky, S. Solid-phase synthesis of molecularly imprinted nanoparticles. Nat. Protoc. 2016, 11, 443–455.
- Brahmbhatt, H.; Poma, A.; Pendergraff, H.M.; Watts, J.K.; Turner, N.W. Improvement of DNA recognition through molecular imprinting: Hybrid oligomer imprinted polymeric nanoparticles (oligoMIP NPs). Biomater. Sci. 2016, 4, 281–287.
- Bedwell, T.S.; Anjum, N.; Ma, Y.; Czulak, J.; Poma, A.; Piletska, E.; Whitcombe, M.J.; Piletsky, S.A. New protocol for optimisation of polymer composition for imprinting of peptides and proteins. RSC Adv. 2019, 9, 27849–27855.
- da Silva, M.S.; Casimiro, T. High Affinity Polymers by Molecular Imprinting for Drug Delivery. In Polymerization; De Souza Gomes, A., Ed.; IntechOpen: London, UK, 2012; pp. 145–162.
- Ayari, M.G.; Kadhirvel, P.; Favetta, P.; Plano, B.; Dejous, C.; Carbonnier, B.; Agrofoglio, L.A. Synthesis of imprinted hydrogel microbeads by inverse Pickering emulsion to controlled release of adenosine 5′-monophosphate. Mater. Sci. Eng. C 2019, 101, 254–263.
- Sedghi, R.; Yassari, M.; Heidari, B. Thermo-responsive molecularly imprinted polymer containing magnetic nanoparticles: Synthesis, characterization and adsorption properties for curcumin. Colloids Surf. B Biointerfaces 2018, 162, 154–162.
- He, S.; Zhang, L.; Bai, S.; Yang, H.; Cui, Z.; Zhang, X.; Li, Y. Advances of molecularly imprinted polymers (MIP) and the application in drug delivery. Eur. Polym. J. 2021, 143, 110179.
- Puoci, F.; Cirillo, G.; Curcio, M.; Parisi, O.I.; Iemma, F.; Picci, N. Molecularly imprinted polymers in drug delivery: State of art and future perspectives. Expert Opin. Drug Deliv. 2011, 8, 1379–1393.
- Liu, Z.; Huang, Y.; Yang, Y. Molecularly Imprinted Polymers as Advanced Drug Delivery Systems. Synthesis, Character and Application, 1st ed.; Springer Nature: Singapore, 2021; p. 216.
- Paul, P.K.; Treetong, A.; Suedee, R. Biomimetic insulin-imprinted polymer nanoparticles as a potential oral drug delivery system. Acta Pharm. 2017, 67, 149–168.
- Bodoki, A.E.; Iacob, B.C.; Bodoki, E. Perspectives of Molecularly Imprinted Polymer-Based Drug Delivery Systems in Cancer Therapy. Polymers 2019, 11, 2085.
- Norell, M.C.; Andersson, H.S.; Nicholls, I.A. Theophylline molecularly imprinted polymer dissociation kinetics: A novel sustained release drug dosage mechanism. J. Mol. Recognit. 1998, 11, 98–102.
- Tuwahatu, C.A.; Yeung, C.C.; Lam, Y.W.; Roy, V.A.L. The molecularly imprinted polymer essentials: Curation of anticancer, ophthalmic, and projected gene therapy drug delivery systems. J. Control. Release 2018, 287, 24–34.
- Li, L.; Chen, L.; Zhang, H.; Yang, Y.; Liu, X.; Chen, Y. Temperature and magnetism bi-responsive molecularly imprinted polymers: Preparation, adsorption mechanism and properties as drug delivery system for sustained release of 5-fluorouracil. Mater. Sci. Eng. C 2016, 61, 158–168.
- Cazares-Cortes, E.; Nerantzaki, M.; Fresnais, J.; Wilhelm, C.; Griffete, N.; Menager, C. Magnetic Nanoparticles Create Hot Spots in Polymer Matrix for Controlled Drug Release. Nanomaterials 2018, 8, 850.
- Zhu, Y.; Liu, R.; Huang, H.; Zhu, Q. Vinblastine-Loaded Nanoparticles with Enhanced Tumor-Targeting Efficiency and Decreasing Toxicity: Developed by One-Step Molecular Imprinting Process. Mol. Pharm. 2019, 16, 2675–2689.
- Minhas, M.U.; Ahmad, M.; Ali, L.; Sohail, M. Synthesis of chemically cross-linked polyvinyl alcohol-co-poly (methacrylic acid) hydrogels by copolymerization; a potential graft-polymeric carrier for oral delivery of 5-fluorouracil. DARU J. Pharm. Sci. 2013, 21, 44.
- Li, F.; Sun, J.; Zhu, H.; Wen, X.; Lin, C.; Shi, D. Preparation and characterization novel polymer-coated magnetic nanoparticles as carriers for doxorubicin. Colloids Surf. B Biointerfaces 2011, 88, 58–62.
- Liechty, W.B.; Peppas, N.A. Expert opinion: Responsive polymer nanoparticles in cancer therapy. Eur. J. Pharm. Biopharm. 2012, 80, 241–246.
- Yoo, J.; Park, C.; Yi, G.; Lee, D.; Koo, H. Active targeting strategies using biological ligands for nanoparticle drug delivery systems. Cancers 2019, 11, 640.
- Bertrand, N.; Wu, J.; Xu, X.; Kamaly, N.; Farokhzad, O.C. Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv. Drug Deliv. Rev. 2014, 66, 2–25.
- Gaitzsch, J.; Delahaye, M.; Poma, A.; Du Prez, F.; Battaglia, G. Comparison of metal free polymer–dye conjugation strategies in protic solvents. Polym. Chem. 2016, 7, 3046–3055.
- Liu, M.; Apriceno, A.; Sipin, M.; Scarpa, E.; Rodriguez-Arco, L.; Poma, A.; Marchello, G.; Battaglia, G.; Angioletti-Uberti, S. Combinatorial entropy behaviour leads to range selective binding in ligand-receptor interactions. Nat. Commun. 2020, 11, 4836.
- Tian, X.; Leite, D.M.; Scarpa, E.; Nyberg, S.; Fullstone, G.; Forth, J.; Matias, D.; Apriceno, A.; Poma, A.; Duro-Castano, A.; et al. On the shuttling across the blood-brain barrier via tubule formation: Mechanism and cargo avidity bias. Sci. Adv. 2020, 6, eabc4397.
- You, J.; Li, X.; de Cui, F.; Du, Y.-Z.; Yuan, H.; Hu, F.Q. Folate-conjugated polymer micelles for active targeting to cancer cells: Preparation, in vitro evaluation of targeting ability and cytotoxicity. Nanotechnology 2008, 19, 045102.
- Zhang, H.; Li, F.; Yi, J.; Gu, C.; Fan, L.; Qiao, Y.; Tao, Y.; Cheng, C.; Wu, H. Folate-decorated maleilated pullulan–doxorubicin conjugate for active tumor-targeted drug delivery. Eur. J. Pharm. Sci. 2011, 42, 517–526.
- Zhu, Y.; Zhang, J.; Meng, F.; Deng, C.; Cheng, R.; Feijen, J.; Zhong, Z. cRGD-functionalized reduction-sensitive shell-sheddable biodegradable micelles mediate enhanced doxorubicin delivery to human glioma xenografts in vivo. J. Control. Release 2016, 233, 29–38.
- Roncato, F.; Rruga, F.; Porcù, E.; Casarin, E.; Ronca, R.; Maccarinelli, F.; Realdon, N.; Basso, G.; Alon, R.; Viola, G. Improvement and extension of anti-EGFR targeting in breast cancer therapy by integration with the Avidin-Nucleic-Acid-Nano-Assemblies. Nat. Commun. 2018, 9, 1–11.
- Jia, C.; Zhang, M.; Zhang, Y.; Ma, Z.B.; Xiao, N.N.; He, X.W.; Li, W.Y.; Zhang, Y.K. Preparation of Dual-Template Epitope Imprinted Polymers for Targeted Fluorescence Imaging and Targeted Drug Delivery to Pancreatic Cancer BxPC-3 Cells. ACS Appl. Mater. Interfaces 2019, 11, 32431–32440.
- Canfarotta, F.; Lezina, L.; Guerreiro, A.; Czulak, J.; Petukhov, A.; Daks, A.; Smolinska-Kempisty, K.; Poma, A.; Piletsky, S.; Barlev, N.A. Specific Drug Delivery to Cancer Cells with Double-Imprinted Nanoparticles against Epidermal Growth Factor Receptor. Nano Lett. 2018, 18, 4641–4646.
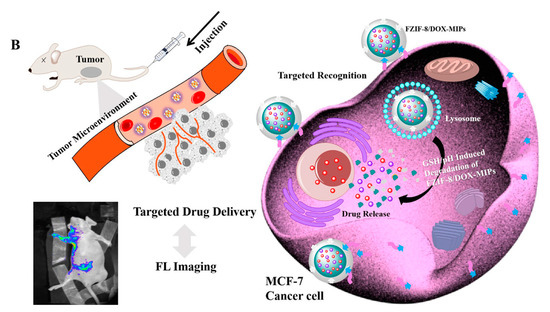
- Qin, Y.T.; Feng, Y.S.; Ma, Y.J.; He, X.W.; Li, W.Y.; Zhang, Y.K. Tumor-Sensitive Biodegradable Nanoparticles of Molecularly Imprinted Polymer-Stabilized Fluorescent Zeolitic Imidazolate Framework-8 for Targeted Imaging and Drug Delivery. ACS Appl. Mater. Interfaces 2020, 12, 24585–24598.
- Cheng, Z.; Al Zaki, A.; Hui, J.Z.; Muzykantov, V.R.; Tsourkas, A. Multifunctional nanoparticles: Cost versus benefit of adding targeting and imaging capabilities. Science 2012, 338, 903–910.
- Fleck, L.M. The costs of caring: Who pays? Who profits? Who panders? Hastings Cent. Rep. 2006, 36, 13–17.
- Das, P.S. Contact Lenses: A Development Towards Ocular Drug Delivery System. World J. Pharm. Res. 2017, 6, 207–216.
- Hilt, J.Z.; Byrne, M.E. Configurational biomimesis in drug delivery: Molecular imprinting of biologically significant molecules. Adv. Drug Deliv. Rev. 2004, 56, 1599–1620.
- Hui, A.; Willcox, M. In vivo studies evaluating the use of contact lenses for drug delivery. Optom. Vis. Sci. 2016, 93, 367–376.
- Bakhshpour, M.; Yavuz, H.; Denizli, A. Controlled release of mitomycin C from PHEMAH-Cu(II) cryogel membranes. Artif. Cells Nanomed. Biotechnol. 2018, 46, 946–954.
- Hassanpour, A.; Irandoust, M.; Soleimani, E.; Zhaleh, H. Increasing the anticancer activity of azidothymidine toward the breast cancer via rational design of magnetic drug carrier based on molecular imprinting technology. Mater. Sci. Eng. C 2019, 103, 109771.
- Mo, C.E.; Chai, M.H.; Zhang, L.P.; Ran, R.X.; Huang, Y.P.; Liu, Z.S. Floating molecularly imprinted polymers based on liquid crystalline and polyhedral oligomeric silsesquioxanes for capecitabine sustained release. Int. J. Pharm. 2019, 557, 293–303.
- Anirudhan, T.S.; Nair, A.S.; Parvathy, J. Extended wear therapeutic contact lens fabricated from timolol imprinted carboxymethyl chitosan-g-hydroxy ethyl methacrylate-g-poly acrylamide as a onetime medication for glaucoma. Eur. J. Pharm. Biopharm. 2016, 109, 61–71.
- Deng, J.; Chen, S.; Chen, J.; Ding, H.; Deng, D.; Xie, Z. Self-Reporting Colorimetric Analysis of Drug Release by Molecular Imprinted Structural Color Contact Lens. ACS Appl. Mater. Interfaces 2018, 10, 34611–34617.
- Mao, C.; Xie, X.; Liu, X.; Cui, Z.; Yang, X.; Yeung, K.W.K.; Pan, H.; Chu, P.K.; Wu, S. The controlled drug release by pH-sensitive molecularly imprinted nanospheres for enhanced antibacterial activity. Mater. Sci. Eng. C 2017, 77, 84–91.
- Vasapollo, G.; Sole, R.D.; Mergola, L.; Lazzoi, M.R.; Scardino, A.; Scorrano, S.; Mele, G. Molecularly imprinted polymers: Present and future prospective. Int. J. Mol. Sci. 2011, 12, 5908–5945.
- Allender, C.; Brain, K.; Heard, C. Molecularly imprinted polymers--preparation, biomedical applications and technical challenges. Prog. Med. Chem. 1999, 36, 235–291.
- He, Y.; Zeng, S.; Abd El-Aty, A.M.; Hacimuftuoglu, A.; Kalekristos Yohannes, W.; Khan, M.; She, Y. Development of Water-Compatible Molecularly Imprinted Polymers Based on Functionalized beta-Cyclodextrin for Controlled Release of Atropine. Polymers 2020, 12, 130.
- Dhanashree, S.; Priyanka, M.; Manisha, K.; Vilasrao, K. Molecularly Imprinted Polymers: Novel Discovery for Drug Delivery. Curr. Drug. Deliv. 2016, 13, 632–645.
- Marcelo, G.; Ferreira, I.C.; Viveiros, R.; Casimiro, T. Development of itaconic acid-based molecular imprinted polymers using supercritical fluid technology for pH-triggered drug delivery. Int. J. Pharm. 2018, 542, 125–131.
- Schweitz, L.; Andersson, L.I.; Nilsson, S. Capillary electrochromatography with predetermined selectivity obtained through molecular imprinting. Anal. Chem. 1997, 69, 1179–1183.
- Striegler, S. Selective discrimination of closely related monosaccharides at physiological pH by a polymeric receptor. Tetrahedron 2001, 57, 2349–2354.
- Grayson, A.C.R.; Choi, I.S.; Tyler, B.M.; Wang, P.P.; Brem, H.; Cima, M.J.; Langer, R. Multi-pulse drug delivery from a resorbable polymeric microchip device. Nat. Mater. 2003, 2, 767–772.
- Farra, R.; Sheppard, N.F.; McCabe, L.; Neer, R.M.; Anderson, J.M.; Santini, J.T.; Cima, M.J.; Langer, R. First-in-human testing of a wirelessly controlled drug delivery microchip. Sci. Transl. Med. 2012, 4, 122ra21.
- Sutradhar, K.B.; Sumi, C.D. Implantable microchip: The futuristic controlled drug delivery system. Drug Deliv. 2016, 23, 1–11.
- Fuchs, Y.; Soppera, O.; Haupt, K. Photopolymerization and photostructuring of molecularly imprinted polymers for sensor applications—A review. Anal. Chim. Acta 2012, 717, 7–20.


