2.3. Active Targeting Drug Delivery
Conventional DDSs usually deliver drugs to both healthy and unhealthy tissues, even though those drugs may damage the healthy cells and thus cause severe side effects, particularly if the drugs are highly toxic (e.g., anti-tumor drugs). Active targeting is another important feature of controlled DDSs, which can spatially control the release of drugs to the target sites within the body, thereby reducing the side effects associated with damage to healthy cells, thus improving the overall therapeutic profile.
The induction of external stimuli to activate the drug delivery vehicles loaded with toxic drugs is an effective method of site-targeting as described above. If external guidance is not used, however, these DDSs will be distributed all over the body.
Therefore, polymeric DDSs with active targeting have been employed to recognize specific cell markers and deliver drugs precisely to the target sites. Active targeting of conventional polymeric DDSs is normally achieved via conjugation with the ligands of specific receptors expressed on target cells. Potential ligands include proteins, peptides, carbohydrates, nucleic acids and small molecules [
129,
130,
131,
132,
133,
134].
Nevertheless, actively targeted polymeric DDSs frequently face complex production processes, significantly high costs (e.g., the RGD peptide targeting sequence costs 145 USD per 10 mg) and/or poor drug release control [
135,
136,
137,
138].
In recent years, double-imprinted polymeric DDSs have been studied for active targeting drug delivery because of the unique features of MIPs as described in the introduction section.
For example, Jia et al. synthesized dual-template silicon MIP NPs used for theranostic applications towards pancreatic cancer BxPC-3 cells that overexpress human fibroblast growth-factor-inducible 14 (FN14) [
139]. The 71–80 peptide of FN14 (FH) and bleomycin were used simultaneously as the template molecules to obtain the active targeting MIP NPs. Optical bioimaging technology makes it possible to use silicon NPs to diagnose cancer. The study showed that bleomycin adsorption to the MIP NPs (>4000 mg g
−1) was 4-fold higher than to the NIP NPs (1100 mg g
−1), suggesting that the loading capacity was markedly enhanced by the imprinting process. The FH adsorption capacity of the MIP NPs (450 mg g
−1) was also higher than that of the NIP NPs (130 mg g
−1), with 2.5-fold higher selectivity for the correct targeting peptide in comparison to a scrambled one. The in vitro release study performed at pH 5.3 (mimicking the tumor microenvironment) highlighted that the MIP NPs sustained the release of a total amount of 1900 mg g
−1 of bleomycin over the course of 72 h, while NIP NPs released the drug more quickly and reached equilibrium (750 mg g
−1) after 10 h. At pH 7.4, NIP NPs and MIP NPs released 800 mg g
−1 and 500 mg g
−1, respectively, after 70 h. The in vivo anti-tumor effect was evaluated in mice via a tail vein injection, and MIP NPs achieved the greatest effect. The tumor volumes of the groups treated with physiological saline, NIP NPs and free bleomycin were respectively 2.3-fold, 1.6-fold and 1.5-fold higher than the group treated with MIP NPs. These results demonstrate that MIP NPs have a superior potential for the treatment of pancreatic cancer relative to NIP NPs and free bleomycin thanks to the MIP system enhanced drug loading, specific recognition and sustained release.
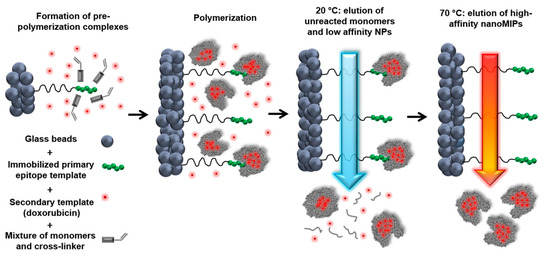
Canfarotta et al. used the solid phase synthesis approach to prepare double-imprinted MIP NPs against the epidermal growth factor receptor (EGFR) that is overexpressed in many types of tumor cells () [
140].
Figure 10. Scheme of the solid-phase synthesis process for double-imprinted nanoMIPs using a peptide epitope of EGFR as primary template attached to the solid phase and doxorubicin as secondary template in solution. Adapted with permission from Canfarotta et al. [
140].
An epitope of the extracellular domain of EGFR and DOX were used as the template molecules. Flow cytometry was used to analyze the selective recognition capability for MIP NPs towards breast cancer cells overexpressing EGFR. Furthermore, DOX-loaded EGFR-MIP NPs decreased the cell viability of EGFR-overexpressing MDA-MB-468 cancer cells (lower than 75%) in comparison to free DOX (more than 90%) and control NPs (~100%). Moreover, the cell viability of non-EGFR-overexpressing cell-lines was virtually unaffected, highlighting the achievement of selective targeting and DOX delivery.
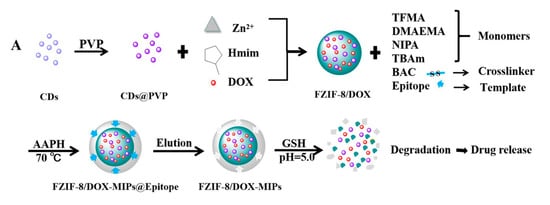
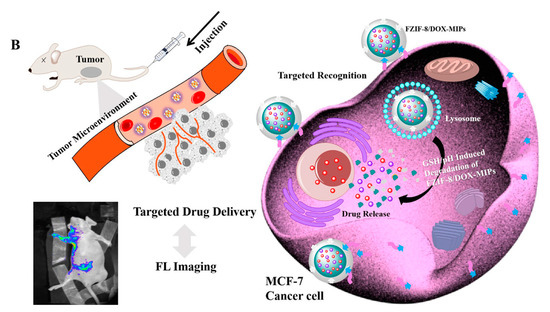
Similarly, Qin et al. studied active targeting MIP NPs for the treatment of breast cancer. The MIP NPs were prepared with an epitope of the CD59 cell membrane glycoprotein and DOX as templates such that the high concentration of GSH and weak acid environment created by the cancer cells would selectively trigger DOX release () [
141].
Figure 11. (
A) Synthesis and GSH/pH dual stimulation degradation route of FZIF-8/DOX-MIPs; (
B) Schematic illustration of targeted imaging and GSH/pH-responsive drug delivery of FZIF-8/DOX-MIPs. Reproduced with permission from Qin et al. [
141].
Fluorescent zeolitic imidazolate framework-8 (FZIF-8) was employed as the framework of the MIP NPs because FZIF-8 is fully biodegradable in an acidic environment. The fluorescence intensity of the MIP NPs was ~4-fold higher than that of the NIP NPs with 0.1 mg mL−1 of the epitope, indicating the enhanced adsorption and strong specificity of the MIP NPs. After 15 days and in absence of GSH, about 50% of the drug leaked from pristine FZIF-8/DOX in PBS (pH 7.4), while almost no DOX was released from FZIF-8/DOX MIP NPs in PBS (at pH 5.0 and 7.4). In the presence of GSH, the DOX release from the MIP NPs greatly increased (at pH 5.0 and 6.0) to more than 90% over 15 days. In addition, the viability of MCF-7 cells (CD59 positive) was significantly lower after treatment with the FZIF-8/DOX MIP NPs (20% cell survival after 72 h exposure to 40 μg mL−1) than after control treatments: FZIF-8/DOX NIP NPs (50%), FZIF-8/DOX (60%) and free DOX (60%). Importantly, normal cells did not show significant apoptosis; the anti-tumor effects were limited to the MCF-7 cancer cells. Moreover, the tumor volume (10 mm) in mice treated with FZIF-8/DOX MIP NPs was more than 2-fold smaller than that in mice treated with the controls. These results indicate that FZIF-8 MIP NPs can selectively target MCF-7 cells and trigger an adequate drug release in the presence of GSH, thereby reducing systemic toxicity and improving the therapeutic index of DOX.
As can be seen, in comparison to the conventional active targeting polymeric DDSs, double-imprinted MIP NPs offer a benefit by significantly reducing aspecific drug release and improving therapeutic effects. In addition, while biological ligands might exhibit superior specificity in certain instances, the high costs and complex production processes associated with materials such as antibodies and peptides may be prohibitive [
131,
142,
143].
Double-imprinted polymeric DDSs, on the other hand, are extremely affordable. Solid-phase synthesis technology, in particular, makes it possible to recycle the templates depending on the production conditions, which greatly optimizes the resources whilst containing the costs [
50]. Furthermore, it is facile to prepare double-imprinted polymeric DDSs in comparison to analogous conjugates [
50,
57,
140].
3. Current Challenges in MIP-Based DDS
In the last 5 years, a myriad of interesting studies has revealed great potential for the development of MIP-based DDSs. Many have focused on drug delivery for anti-tumor agents, which are important candidates for improved DDSs because the majority of these drugs possess a narrow therapeutic window. Methacrylic monomers and cross-linkers such as HEMA, MAA, MBA, EGDMA and EDMA have been widely used in the advancement of MIP-based DDSs [
144,
145]. These monomers are considered to have acceptable toxicity and excellent biocompatibility [
78,
120,
127], though their long-term toxicity and metabolic pathways have not been evaluated in depth. Besides, additional materials such as POSS [
45,
104], β-CD [
45], CS [
89], silicon [
91,
139] and stimuli-sensitive materials [
47,
70,
124] have been used to modify MIPs to improve performance parameters such as drug release behavior, biodegradability and biocompatibility [
67].
Another major obstacle in the development of most MIP-based DDSs is the use of organic solvents (e.g., toluene, chloroform) [
67]. These facilitate and maintain the non-covalent interactions between the template molecules and the functional monomers [
146,
147]. Nonetheless, in terms of medical translation and applications, the presence of residual organic solvents can damage healthy cells, leading to serious side effects [
146,
148]. Furthermore, the use of organic solvents during synthesis may result in a marked difference in drug-release behavior of MIPs in aqueous media, as well as potentially increasing the manufacturing costs [
148,
149]. Alternatives such as supercritical carbon dioxide (scCO2) technology exhibit great environmental and safety advantages because of the unique and beneficial features of the prepared polymers, such as a controlled morphology, non-toxicity, absence of solvent residues [
60] and the ability to avoid purification and drying steps [
150]. Furthermore, scCO2 can stabilize hydrogen bonds between the templates and functional monomers [
151]. The technology, though, is still not widely used nor easily accessible (even at a laboratory scale), possibly due to the need for specialized expensive equipment as well as the extreme operational parameters (temperatures and pressures). Nonetheless, scCO
2 has a great potential for the development of highly pure, GMP-compliant MIP-based DDSs [
150].
A further challenge that arises from the analysis of the above-discussed literature examples, is that only a limited number of studies reported in vivo data for the evaluation of MIP-based DDSs. In vivo research should be considered an indispensable step in the evaluation of any potential DDS. Since most studies evaluated toxicity and drug release only in vitro, further investigations and data are needed to ensure that the promising in vitro behavior of MIPs translates in in vivo models exhibiting safety and adequate release properties. Only through in vivo studies can we adequately evaluate the possible advantages of MIPs in clinical practice.
Another relevant concern in some cases is the inadequate drug loading of MIP-based DDSs (e.g., in contact lenses). This may result in sub-par, ineffective drug release. Importantly, though, even MIP-based DDSs with somewhat inadequate loading usually perform better than the corresponding NIP-based DDSs, indicating the potential advantages of molecular imprinting to enhance drug loading. The loading capacity may be increased by employing a smaller proportion of cross-linkers, but this strategy must also consider the trade-off between drug loading and the stability of imprinted cavities.
Furthermore, some MIPs may undergo a small initial burst release due to the adsorption of drug molecules onto their surface. For some drugs (e.g., anti-tumor agents), this may cause serious toxic effects. In other cases, however, burst release can be beneficial (e.g., at the beginning of antibiotic treatment) [
42]. Perhaps better purification strategies can allow achieving an improved modulation of the burst effect. Although it is tremendously challenging to design a suitable release profile, MIPs offer ample opportunities for modifications that can implement a variety of release profiles to fit the requirements of the drug of interest.
4. Conclusions and Future Perspectives
Conventional drug delivery can be burdened by systemic toxicity and low bioavailability because of non-specific delivery and rapid clearance. Recently, in attempts to address these limitations, a variety of MIPs have been evaluated as novel DDSs. An analysis of the literature on MIP-based DDSs reveals a great potential for extensive research, particularly for the delivery of drugs with narrow therapeutic windows and/or low bioavailability. Although the process of designing and translating MIP-based DDSs into clinical practice is still in its infancy, ongoing development will most likely lead to the creation of innovative drug delivery vehicles with commercial value.
Imprinted polymers can not only significantly enhance the drug loading and the stability of drugs in harsh conditions but also attenuate the release behavior by engineering specific interactions between drug molecules and functional monomers. As an example of the advantages of rate-programmed drug delivery, MIPs as excipients can be used to develop sustained transdermal formulations, therapeutic contact lenses and oral formulations for protein delivery. In addition, MIP-based DDSs can be designed with stimuli-responsiveness, using properties of the imprinted cavities to achieve enhanced release profiles compared to conventional polymeric DDSs. Even more interestingly, active targeting drug delivery can be achieved via unique double-imprinting of targeting moieties and drugs.
However, much of the extant research does not go so far as to include clinical studies that address biomedical regulations regarding the novel drug delivery devices. Although some studies report the in vitro cell toxicity of MIP-based DDSs, the therapeutic effects and clinical safety cannot be determined without in vivo assessments. Therefore, additional ongoing efforts are needed to design, develop and evaluate MIP-based DDSs to evaluate their safety profiles and satisfy biomedical regulations.
With an eye on the horizon, a combination of MIP-based DDSs and implantable microchips has great potential for self-regulated drug delivery [
5] as the novel DDS can detect changes in the level of a biochemical substance (e.g., glucose) and can prompt the rapid release of the drug (e.g., insulin) under the desired conditions [
3]. Striegler proposed a MIP-based α-glucose-biosensor in which pH changes in response to changes in the glucose concentration in the environment [
152]. In addition, several interesting studies have achieved controlled drug delivery based on implantable microchips [
153,
154,
155]. Therefore, the hybrid of the MIP and implantable microchips may prove to be a promising drug delivery avenue within reach of clinical applications in the coming years [
5,
156].