 +1 credit
+1 credit
Video Upload Options
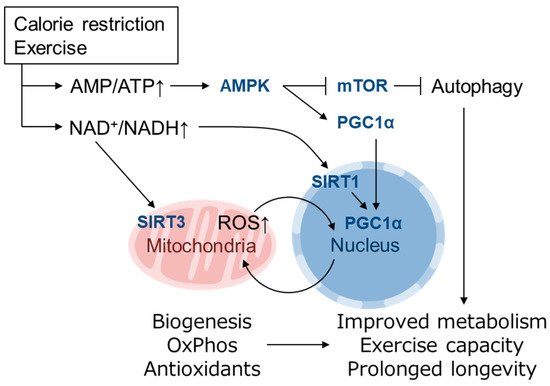
Reactive oxygen species (ROS) are byproducts of aerobic respiration and signaling molecules that control various cellular functions. Nrf2 governs the gene expression of endogenous antioxidant synthesis and ROS-eliminating enzymes in response to various electrophilic compounds that inactivate the negative regulator Keap1. Accumulating evidence has shown that mitochondrial ROS (mtROS) activate Nrf2, often mediated by certain protein kinases, and induce the expression of antioxidant genes and genes involved in mitochondrial quality/quantity control. Mild physiological stress, such as caloric restriction and exercise, elicits beneficial effects through a process known as “mitohormesis”. Exercise induces NOX4 expression in the heart, which activates Nrf2 and increases endurance capacity. Mice transiently depleted of SOD2 or overexpressing skeletal muscle-specific UCP1 exhibit Nrf2-mediated antioxidant gene expression and PGC1α-mediated mitochondrial biogenesis. ATF4 activation may induce a transcriptional program that enhances NADPH synthesis in the mitochondria and might cooperate with the Nrf2 antioxidant system. In response to severe oxidative stress, Nrf2 induces Klf9 expression, which represses mtROS-eliminating enzymes to enhance cell death. Nrf2 is inactivated in certain pathological conditions, such as diabetes, but Keap1 down-regulation or mtROS elimination rescues Nrf2 expression and improves the pathology. These reports aid us in understanding the roles of Nrf2 in pathophysiological alterations involving mtROS.
1. Introduction
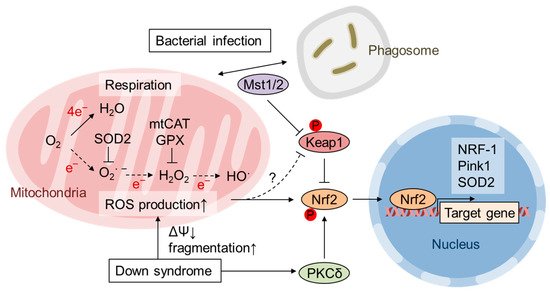
2. Activation of Nrf2 by mtROS
3. Role of mtROS and Nrf2 in Mitohormesis
References
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313.
- Lane, N. Mitonuclear match: Optimizing fitness and fertility over generations drives ageing within generations. Bioessays 2011, 33, 860–869.
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418.
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748.
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell. Physiol. Biochem. 2017, 44, 532–553.
- Li, H.; Forstermann, U. Uncoupling of endothelial NO synthase in atherosclerosis and vascular disease. Curr. Opin. Pharm. 2013, 13, 161–167.
- Battelli, M.G.; Polito, L.; Bortolotti, M.; Bolognesi, A. Xanthine oxidoreductase-derived reactive species: Physiological and pathological effects. Oxidative Med. Cell. Longev. 2016, 2016, 3527579.
- Mitsuishi, Y.; Motohashi, H.; Yamamoto, M. The Keap1-Nrf2 system in cancers: Stress response and anabolic metabolism. Front. Oncol. 2012, 2, 200.
- Itoh, K.; Ye, P.; Matsumiya, T.; Tanji, K.; Ozaki, T. Emerging functional cross-talk between the Keap1-Nrf2 system and mitochondria. J. Clin. Biochem. Nutr. 2015, 56, 91–97.
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203.
- Forman, H.J.; Davies, K.J.; Ursini, F. How do nutritional antioxidants really work: Nucleophilic tone and para-hormesis versus free radical scavenging In Vivo. Free Radic. Biol. Med. 2014, 66, 24–35.
- Wang, Y.; Branicky, R.; Noe, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928.
- Cox, C.S.; McKay, S.E.; Holmbeck, M.A.; Christian, B.E.; Scortea, A.C.; Tsay, A.J.; Newman, L.E.; Shadel, G.S. Mitohormesis in Mice via Sustained Basal Activation of Mitochondrial and Antioxidant Signaling. Cell Metab. 2018, 28, 776–786.
- Zamponi, E.; Zamponi, N.; Coskun, P.; Quassollo, G.; Lorenzo, A.; Cannas, S.A.; Pigino, G.; Chialvo, D.R.; Gardiner, K.; Busciglio, J.; et al. Nrf2 stabilization prevents critical oxidative damage in Down syndrome cells. Aging Cell 2018, 17, e12812.
- Wang, P.; Geng, J.; Gao, J.; Zhao, H.; Li, J.; Shi, Y.; Yang, B.; Xiao, C.; Linghu, Y.; Sun, X.; et al. Macrophage achieves self-protection against oxidative stress-induced ageing through the Mst-Nrf2 axis. Nat. Commun. 2019, 10, 755.
- Piantadosi, C.A.; Carraway, M.S.; Babiker, A.; Suliman, H.B. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ. Res. 2008, 103, 1232–1240.
- Murata, H.; Takamatsu, H.; Liu, S.; Kataoka, K.; Huh, N.H.; Sakaguchi, M. NRF2 Regulates PINK1 Expression under oxidative stress conditions. PLoS ONE 2015, 10, e0142438.
- Ryoo, I.G.; Kwak, M.K. Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria. Toxicol. Appl. Pharm. 2018, 359, 24–33.
- Cherry, A.D.; Suliman, H.B.; Bartz, R.R.; Piantadosi, C.A. Peroxisome proliferator-activated receptor gamma co-activator 1-α as a critical co-activator of the murine hepatic oxidative stress response and mitochondrial biogenesis in Staphylococcus aureus sepsis. J. Biol. Chem. 2014, 289, 41–52.
- Hinchy, E.C.; Gruszczyk, A.V.; Willows, R.; Navaratnam, N.; Hall, A.R.; Bates, G.; Bright, T.P.; Krieg, T.; Carling, D.; Murphy, M.P. Mitochondria-Derived ROS activate AMP-activated protein kinase (AMPK) indirectly. J. Biol. Chem. 2018, 293, 17208–17217.
- Davies, K.J. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life 2000, 50, 279–289.
- Jovaisaite, V.; Mouchiroud, L.; Auwerx, J. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J. Exp. Biol. 2014, 217, 137–143.
- Tsushima, M.; Liu, J.; Hirao, W.; Yamazaki, H.; Tomita, H.; Itoh, K. Emerging evidence for crosstalk between Nrf2 and mitochondria in physiological homeostasis and in heart disease. Arch. Pharm. Res. 2019, 1–11.
- Khan, A.U.H.; Allende-Vega, N.; Gitenay, D.; Garaude, J.; Vo, D.N.; Belkhala, S.; Gerbal-Chaloin, S.; Gondeau, C.; Daujat-Chavanieu, M.; Delettre, C.; et al. Mitochondrial Complex I activity signals antioxidant response through ERK5. Sci. Rep. 2018, 8, 7420.
- Indo, H.P.; Hawkins, C.L.; Nakanishi, I.; Matsumoto, K.I.; Matsui, H.; Suenaga, S.; Davies, M.J.; St Clair, D.K.; Ozawa, T.; Majima, H.J. Role of mitochondrial reactive oxygen species in the activation of cellular signals, molecules, and function. Handb. Exp. Pharm. 2017, 240, 439–456.
- Robb, E.L.; Gawel, J.M.; Aksentijevic, D.; Cocheme, H.M.; Stewart, T.S.; Shchepinova, M.M.; Qiang, H.; Prime, T.A.; Bright, T.P.; James, A.M.; et al. Selective superoxide generation within mitochondria by the targeted redox cycler MitoParaquat. Free Radic. Biol. Med. 2015, 89, 883–894.
- Imhoff, B.R.; Hansen, J.M. Extracellular redox status regulates Nrf2 activation through mitochondrial reactive oxygen species. Biochem. J. 2009, 424, 491–500.
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; Van Remmen, H.; et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308, 1909–1911.
- Treuting, P.M.; Linford, N.J.; Knoblaugh, S.E.; Emond, M.J.; Morton, J.F.; Martin, G.M.; Rabinovitch, P.S.; Ladiges, W.C. Reduction of age-associated pathology in old mice by overexpression of catalase in mitochondria. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2008, 63, 813–822.
- Lee, H.Y.; Choi, C.S.; Birkenfeld, A.L.; Alves, T.C.; Jornayvaz, F.R.; Jurczak, M.J.; Zhang, D.; Woo, D.K.; Shadel, G.S.; Ladiges, W.; et al. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab. 2010, 12, 668–674.
- Mao, P.; Manczak, M.; Calkins, M.J.; Truong, Q.; Reddy, T.P.; Reddy, A.P.; Shirendeb, U.; Lo, H.H.; Rabinovitch, P.S.; Reddy, P.H. Mitochondria-Targeted catalase reduces abnormal APP processing, amyloid beta production and BACE1 in a mouse model of Alzheimer’s disease: Implications for neuroprotection and lifespan extension. Hum. Mol. Genet. 2012, 21, 2973–2990.
- Umanskaya, A.; Santulli, G.; Xie, W.; Andersson, D.C.; Reiken, S.R.; Marks, A.R. Genetically enhancing mitochondrial antioxidant activity improves muscle function in aging. Proc. Natl. Acad. Sci. USA 2014, 111, 15250–15255.
- Geng, J.; Sun, X.; Wang, P.; Zhang, S.; Wang, X.; Wu, H.; Hong, L.; Xie, C.; Li, X.; Zhao, H.; et al. Kinases Mst1 and Mst2 positively regulate phagocytic induction of reactive oxygen species and bactericidal activity. Nat. Immunol. 2015, 16, 1142–1152.
- Vina, J.; Borras, C.; Abdelaziz, K.M.; Garcia-Valles, R.; Gomez-Cabrera, M.C. The free radical theory of aging revisited: The cell signaling disruption theory of aging. Antioxid. Redox Signal. 2013, 19, 779–787.
- Tapia, P.C. Sublethal mitochondrial stress with an attendant stoichiometric augmentation of reactive oxygen species may precipitate many of the beneficial alterations in cellular physiology produced by caloric restriction, intermittent fasting, exercise and dietary phytonutrients: “Mitohormesis” for health and vitality. Med. Hypotheses 2006, 66, 832–843.
- Palmeira, C.M.; Teodoro, J.S.; Amorim, J.A.; Steegborn, C.; Sinclair, D.A.; Rolo, A.P. Mitohormesis and metabolic health: The interplay between ROS, cAMP and sirtuins. Free Radic. Biol. Med. 2019, 141, 483–491.
- Barcena, C.; Mayoral, P.; Quiros, P.M. Mitohormesis, an antiaging paradigm. Int. Rev. Cell Mol. Biol. 2018, 340, 35–77.
- Quirós, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213–226.
- Lee, S.-H.; Lee, J.-H.; Lee, H.-Y.; Min, K.-J. Sirtuin signaling in cellular senescence and aging. BMB Rep. 2019, 52, 24–34.
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580.
- Sinclair, D.A.; Guarente, L. Extrachromosomal rDNA circles—A cause of aging in yeast. Cell 1997, 91, 1033–1042.
- Tissenbaum, H.A.; Guarente, L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 2001, 410, 227–230.
- Wood, J.G.; Rogina, B.; Lavu, S.; Howitz, K.; Helfand, S.L.; Tatar, M.; Sinclair, D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 2004, 430, 686–689.
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S.
- Tan, Z.; Luo, X.; Xiao, L.; Tang, M.; Bode, A.M.; Dong, Z.; Cao, Y. The role of PGC1α in cancer metabolism and its therapeutic implications. Mol. Cancer 2016, 15, 774–782.
- Qian, X.; Li, X.; Shi, Z.; Bai, X.; Xia, Y.; Zheng, Y.; Xu, D.; Chen, F.; You, Y.; Fang, J.; et al. KDM3A senses oxygen availability to regulate PGC-1α-mediated mitochondrial biogenesis. Mol. Cell 2019, 76, 885–895.
- Lamming, D.W. Inhibition of the mechanistic target of rapamycin (mTOR)-rapamycin and beyond. Cold Spring Harb. Perspect. Med. 2016, 6.
- Warburton, D.E.; Nicol, C.W.; Bredin, S.S. Health benefits of physical activity: The evidence. CMAJ 2006, 174, 801–809.
- Lanza, I.R.; Short, D.K.; Short, K.R.; Raghavakaimal, S.; Basu, R.; Joyner, M.J.; McConnell, J.P.; Nair, K.S. Endurance exercise as a countermeasure for aging. Diabetes 2008, 57, 2933–2942.
- Vargas-Ortiz, K.; Perez-Vazquez, V.; Macias-Cervantes, M.H. Exercise and sirtuins: A way to mitochondrial health in skeletal muscle. Int. J. Mol. Sci. 2019, 20, 2717.
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766.
- Martinus, R.D.; Garth, G.P.; Webster, T.L.; Cartwright, P.; Naylor, D.J.; Hoj, P.B.; Hoogenraad, N.J. Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur. J. Biochem. 1996, 240, 98–103.
- Munch, C.; Harper, J.W. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 2016, 534, 710–713.
- Munch, C. The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol. 2018, 16, 81.
- Blackwell, T.K.; Steinbaugh, M.J.; Hourihan, J.M.; Ewald, C.Y.; Isik, M. SKN-1/Nrf, stress responses, and aging in Caenorhabditis elegans. Free Radic. Biol. Med. 2015, 88, 290–301.
- Pulliam, D.A.; Deepa, S.S.; Liu, Y.; Hill, S.; Lin, A.L.; Bhattacharya, A.; Shi, Y.; Sloane, L.; Viscomi, C.; Zeviani, M.; et al. Complex IV-deficient Surf1(-/-) mice initiate mitochondrial stress responses. Biochem. J. 2014, 462, 359–371.
- Wedatilake, Y.; Brown, R.M.; McFarland, R.; Yaplito-Lee, J.; Morris, A.A.; Champion, M.; Jardine, P.E.; Clarke, A.; Thorburn, D.R.; Taylor, R.W.; et al. SURF1 deficiency: A multi-centre natural history study. Orphanet J. Rare Dis. 2013, 8, 96.
- Dell’agnello, C.; Leo, S.; Agostino, A.; Szabadkai, G.; Tiveron, C.; Zulian, A.; Prelle, A.; Roubertoux, P.; Rizzuto, R.; Zeviani, M. Increased longevity and refractoriness to Ca2+-dependent neurodegeneration in Surf1 knockout mice. Hum. Mol. Genet. 2007, 16, 431–444.


