 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maria Dalamaga | + 5219 word(s) | 5219 | 2021-06-28 08:37:32 | | | |
| 2 | Bruce Ren | -21 word(s) | 5198 | 2021-06-28 10:20:28 | | |
Video Upload Options
Although comprising a much smaller proportion of the human microbiome, the fungal community has gained much more attention lately due to its multiple and yet undiscovered interactions with the human bacteriome and the host. Head and neck cancer carcinoma, colorectal carcinoma, and pancreatic ductal adenocarcinoma have been associated with dissimilarities in the composition of the mycobiome between cases with cancer and non-cancer subjects. In particular, an abundance of Malassezia has been associated with the onset and progression of colorectal carcinoma and pancreatic adenocarcinoma, while the genera Schizophyllum, a member of the oral mycobiome, is suggested to exhibit anti-cancer potential. The use of multi-omics will further assist in establishing whether alterations in the human mycobiome are causal or a consequence of specific types of cancers.
1. Introduction
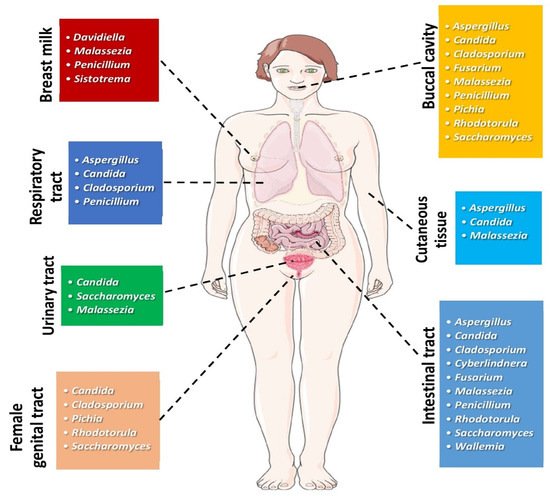
2. Mycobiome and Head and Neck Cancer
| Research/Year | Population, Type of Study | Clinical Specimen | Main Findings | Remarks |
|---|---|---|---|---|
| Head and Neck Cancer | ||||
| Perera et al., 2017 [26] | 52 individuals; 25 with OSCC; 27 intra-oral-fibro epithelial polyps | 52 biopsies from 25 patients with OSCC and 27 with oral polyps. DNA was extracted and sequenced for the ITS2 region | 364 species accounting for 160 genera and 2 phyla (Ascomycota and Basidiomycota) were detected. Candida and Malassezia made up 48% and 11% of the average fungal community, respectively, according to Luan et al., 2015. |
-5 species and 4 genera were identified in more than half of samples. -Less abundance and diversity in OSCC tissues of patients. -Candida, Hannaella, and Gibberella were ↑↑ in OSCC; Altenaria and Trametes were in greater quantity in polyps specimens. -Candida albicans, Candida etchellsii, and Hannaella luteola–like species were enriched in OSCC Hanseniaspora uvarum–like species, Malassezia restricta, and Aspergillus tamarii are predominant in polyps specimens. -Dysbiotic mycobiome dominated by C. albicans has been observed in OSCC. |
| Mukherjee et al., 2017 [25] | 39 participants with OSCC of the tongue | 39 tissue samples from oral SCC and adjacent tissues were analyzed after DNA extraction for 16S/18S rRNA gene. | Fungal richness was ↓↓ in tumor tissue (TT) in comparison to the adjacent non-cancerous tissue (ANCT), p < 0.006. The presence of 22 bacterial and 7 fungal genera was different in TT and ANCT. Aspergillus in TT was negatively associated with the presence of bacteria Actinomyces, Prevotella, Streptococcus, whilst it presented a positive association with Aggregatibacter. |
-Subjects with advanced T-stage disease presented reduced mean differences between TT and ANCT, in comparison to subjects with regional disease. -Findings indicative of differences in the bacteriome and mycobiome between OSCC patients and their adjacent non-cancerous oral epithelium -Association with T-stage. -Despite the similarities in the index of diversity of the mycobiome between TT and ANCT, the abundance of the mycobiome was diminished in TT. -This study is suggestive of existing changes in the local environment in patients with OSCC, expressed as specific bacterial and fungal dysbiosis |
| Vesty et al., 2018 [27] | 30 participants, including 14 patients with HNSCC | Saliva specimens analyzed by 16S rRNA gene and ITS1amplicon sequencing | ↑↑ Candida Candida albicans representing more than 96% of fungi in the majority of subjects with HNSCC. |
-↑↑ IL-1β and IL-8 in HNSCC and patients with poor dental health, when compared to healthy controls. -IL-1β and IL-8 levels were associated with C. albicans. -In HNSCC, salivary microbial and inflammatory markers are affected by oral hygiene. |
| Shay et al., 2020 [24] | 92 individuals, including 46 patients with HNSCC | Oral wash samples analyzed by 16S rRNA and ITS gene sequencing | Distinct strains of Candida albicans are increased or decreased in oral wash specimens from patients with HNSCC, when compared to healthy controls. | -Distinct strains of Candida albicans and Rothia mucilaginosa differed in numbers. Schizophyllum commune was decreased in HNSCC patients, in comparison to healthy controls. -Compared to controls, oral cavity of subjects with HNSCC presents distinct differences in the mycobiome and bacteriome, and their interactions. |
| Colorectal Cancer | ||||
| Luan et al., 2015 [40] | 27 patients with colorectal adenomas | Biopsies from colorectal adenomas and adjacent tissues were studied by using denaturing gradient gel electrophoresis (DGGE) | ↑↑ Ascomycota, Glomeromycota and Basidiomycota. ↓↓ diversity in adenomas compared to adjacent tissue |
-↑↑ Basidiomycota in adjacent tissues. -↑↑ Basidiomycota and Saccharomycetales in advanced adenoma samples, when compared to non-advanced. |
| Gao et al., 2017 [41] | 131 individuals with colorectal carcinoma (CRC), colorectal polyps and normal subjects | Stool samples from patients with CRC, polyps and normal subjects were analyzed by using ITS2 gene sequencing | ↑ ↑ Ascomycota followed by Basidiomycota ↓↓ diversity in the polyp group, when compared to controls. |
↑↑ Ratio of Ascomycota to Basidiomycota in subjects with CRC and polyps, in comparison to controls. ↑↑ of the opportunistic fungi Trichosporon and Malassezia, which could be implicated in the progression to CRC. |
| Richard et al., 2018 [42] | 27 patients with CRC; 7 with colitis-associated cancer, 10 patients with sporadic cancer and 10 healthy individuals | Tissue specimens from colonic resections in colitis-associated malignancy and sporadic CRC groups were analyzed using 16S rRNA and ITS2 sequencing | ↑↑ Basidiomycota followed by Ascomycota ↓ diversity in sporadic cancer. |
↑↑ Basidiomycota in colitis-associated cancer. |
| Coker et al., 2019 [43] | 585 individuals; 184 patients with CRC, 197 patients colorectal adenomas and 204 normal subjects | Stool samples from patients with CRC, colorectal adenomas and normal subjects were analyzed by fecal shotgun metagenomic sequencing | -Ascomycota, Basidiomycota and Mucoromycota in patients with CRC and healthy participants. -No difference in diversity |
-↑↑ Basidiomycota/Ascomycota ratio in CRC when compared to controls. -14 fungi identified with differential composition between CRC and controls. |
| Pancreatic Cancer | ||||
| Aykut et al., 2019 [44] | (1) Experiments in mice as well as in humans using 18S rRNA sequencing KC mice, which develop spontaneous pancreatic cancer by targeted expression of mutant Kras. C57BL/6, MBL-null, and C3−/− mice. (2) Human stool samples and pancreatic tissue specimens were gathered from healthy volunteers and subjects undergoing surgery for PDA or benign pancreatic disorder. |
Because of the direct proximity and relationship of the intestinal and pancreatic duct via the Oddi sphincter, gut fungi could enter the pancreas. To examine this hypothesis, they administered GFP-labeled Saccharomyces cerevisiae to controls or cancer-bearing mice through oral gavage. Fungi moved into the pancreas in less than thirty minutes, suggesting that the intestinal fungal community may directly impact on the pancreatic microenvironment. | -PDA tumors harbored a ~3000-fold augmentation in fungi, in comparison to physiologic pancreas in both mice and humans. -PDA mycobiome was different from gut or physiologic pancreatic mycobiome based on diversity indexes. -The fungal community infiltrating PDA was ↑↑ enriched in Malassezia in mice and humans. -Fungal elimination with the use of amphotericin B was tumor-protective in slowly progressive as well as in models of invasive PDA, whereas re-population with Malassezia but not Candida, Saccharomyces, or Aspergillus–promoted oncogenesis. |
-Connection of mannose-binding lectin (MBL), that attaches fungal wall glycans to activate the complement pathway, was needed in the promotion of malignancy. -MBL or C3 deletion in the extra-tumoral area or C3aR knockdown in tumor cells prevented tumor expansion. Reprogramming of the fungal ecosystem did not change PDA progression in MBL or C3 deficient mice. -Pathogenic fungi may promote PDA by activating the complement pathway via MBL induction. |
3. Mycobiome and Colorectal Cancer (CRC)
3.1. The Role of Fungal Dysbiosis in CRC
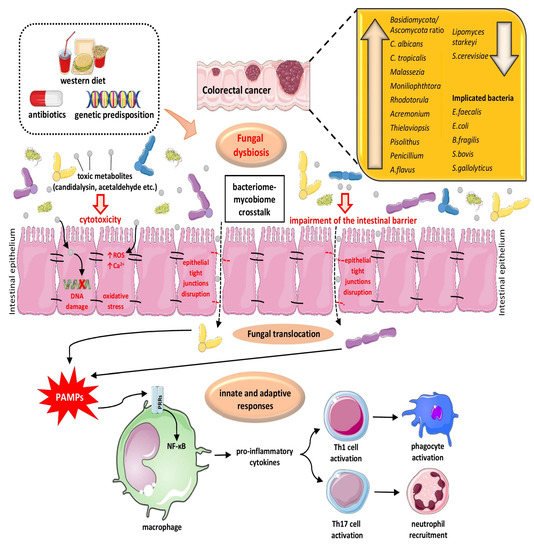
3.2. The Interplay of Gut Microbiome and Mycobiome in Colon Physiology/Pathology and CRC Pathogenesis
4. Mycobiome and Pancreatic Cancer
References
- Nilsson, R.H.; Anslan, S.; Bahram, M.; Wurzbacher, C.; Baldrian, P.; Tedersoo, L. Mycobiome diversity: High-throughput sequencing and identification of fungi. Nat. Rev. Microbiol. 2019, 17, 95–109.
- Cui, L.; Morris, A.; Ghedin, E. The human mycobiome in health and disease. Genome Med. 2013, 5, 63.
- Huffnagle, G.B.; Noverr, M.C. The emerging world of the fungal microbiome. Trends Microbiol. 2013, 21, 334–341.
- Seed, P.C. The human mycobiome. Cold Spring Harb. Perspect. Med. 2014, 5, a019810.
- Vallianou, N.G.; Geladari, E.; Kounatidis, D. Microbiome and hypertension: Where are we now? J. Cardiovasc. Med. 2020, 21, 83–88.
- Auchtung, T.A.; Fofanova, T.Y.; Stewart, C.J.; Nash, A.K.; Wong, M.C.; Gesell, J.R.; Auchtung, J.M.; Ajami, N.J.; Petrosino, J.F. Investigating Colonization of the Healthy Adult Gastrointestinal Tract by Fungi. mSphere 2018, 3.
- Hallen-Adams, H.E.; Suhr, M.J. Fungi in the healthy human gastrointestinal tract. Virulence 2017, 8, 352–358.
- Kong, H.H.; Morris, A. The emerging importance and challenges of the human mycobiome. Virulence 2017, 8, 310–312.
- Ward, T.L.; Dominguez-Bello, M.G.; Heisel, T.; Al-Ghalith, G.; Knights, D.; Gale, C.A. Development of the Human Mycobiome over the First Month of Life and across Body Sites. mSystems 2018, 3.
- Pareek, S.; Kurakawa, T.; Das, B.; Motooka, D.; Nakaya, S.; Rongsen-Chandola, T.; Goyal, N.; Kayama, H.; Dodd, D.; Okumura, R.; et al. Comparison of Japanese and Indian intestinal microbiota shows diet-dependent interaction between bacteria and fungi. NPJ Biofilms Microbiomes 2019, 5, 37.
- Cohen, R.; Roth, F.J.; Delgado, E.; Ahearn, D.G.; Kalser, M.H. Fungal flora of the normal human small and large intestine. N. Engl. J. Med. 1969, 280, 638–641.
- Drgona, L.; Khachatryan, A.; Stephens, J.; Charbonneau, C.; Kantecki, M.; Haider, S.; Barnes, R. Clinical and economic burden of invasive fungal diseases in Europe: Focus on pre-emptive and empirical treatment of Aspergillus and Candida species. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 7–21.
- Li, C.C.; Shen, Z.; Bavarian, R.; Yang, F.; Bhattacharya, A. Oral Cancer: Genetics and the Role of Precision Medicine. Dent. Clin. N. Am. 2018, 62, 29–46.
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Emfietzoglou, R.; Spyrou, N.; Mantzoros, C.S.; Dalamaga, M. Could the endocrine disruptor bisphenol-A be implicated in the pathogenesis of oral and oropharyngeal cancer? Metabolic considerations and future directions. Metabolism 2019, 91, 61–69.
- Ghannoum, M.A.; Jurevic, R.J.; Mukherjee, P.K.; Cui, F.; Sikaroodi, M.; Naqvi, A.; Gillevet, P.M. Characterization of the oral fungal microbiome (mycobiome) in healthy individuals. PLoS Pathog. 2010, 6, e1000713.
- Shelburne, S.A.; Ajami, N.J.; Chibucos, M.C.; Beird, H.C.; Tarrand, J.; Galloway-Peña, J.; Albert, N.; Chemaly, R.F.; Ghantoji, S.S.; Marsh, L.; et al. Implementation of a Pan-Genomic Approach to Investigate Holobiont-Infecting Microbe Interaction: A Case Report of a Leukemic Patient with Invasive Mucormycosis. PLoS ONE 2015, 10, e0139851.
- Mukherjee, P.K.; Hoarau, G.; Gower-Rousseau, C.; Retuerto, M.; Neut, C.; Vermeire, S.; Clemente, J.; Colombel, J.; Poulain, D.; Sendid, B.; et al. Gut Bacteriome (GB) and Mycobiome (GM) in Crohn’s Disease (CD): Association Between Candida tropicalis (CT) and CD (Oral Presentation). In Proceedings of the 2015 Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC)/International Congress of Chemotherapy and Infection (ICC), San Diego, CA, USA, 17–21 September 2015; American Society of Microbiology (ASM): Washington, DC, USA; International Society of Chemotherapy (ISC): Washington, DC, USA, 2015.
- Han, Y.W.; Wang, X. Mobile microbiome: Oral bacteria in extra-oral infections and inflammation. J. Dent. Res. 2013, 92, 485–491.
- Moritani, K.; Takeshita, T.; Shibata, Y.; Ninomiya, T.; Kiyohara, Y.; Yamashita, Y. Acetaldehyde production by major oral microbes. Oral Dis. 2015, 21, 748–754.
- Marttila, E.; Bowyer, P.; Sanglard, D.; Uittamo, J.; Kaihovaara, P.; Salaspuro, M.; Richardson, M.; Rautemaa, R. Fermentative 2-carbon metabolism produces carcinogenic levels of acetaldehyde in Candida albicans. Mol. Oral Microbiol. 2013, 28, 281–291.
- Tillonen, J.; Homann, N.; Rautio, M.; Jousimies-Somer, H.; Salaspuro, M. Role of yeasts in the salivary acetaldehyde production from ethanol among risk groups for ethanol-associated oral cavity cancer. Alcohol Clin. Exp. Res. 1999, 23, 1409–1415.
- Gibson, G.R.; Roberfroid, M.B. Dietary modulation of the human colonic microbiota: Introducing the concept of prebiotics. J. Nutr. 1995, 125, 1401–1412.
- Shay, E.; Sangwan, N.; Padmanabhan, R.; Lundy, S.; Burkey, B.; Eng, C. Bacteriome and mycobiome and bacteriome-mycobiome interactions in head and neck squamous cell carcinoma. Oncotarget 2020, 11, 2375–2386.
- Mukherjee, P.K.; Wang, H.; Retuerto, M.; Zhang, H.; Burkey, B.; Ghannoum, M.A.; Eng, C. Bacteriome and mycobiome associations in oral tongue cancer. Oncotarget 2017, 8, 97273–97289.
- Perera, M.; Al-Hebshi, N.N.; Perera, I.; Ipe, D.; Ulett, G.C.; Speicher, D.J.; Chen, T.; Johnson, N.W. A dysbiotic mycobiome dominated by Candida albicans is identified within oral squamous-cell carcinomas. J. Oral Microbiol. 2017, 9, 1385369.
- Vesty, A.; Gear, K.; Biswas, K.; Radcliff, F.J.; Taylor, M.W.; Douglas, R.G. Microbial and inflammatory-based salivary biomarkers of head and neck squamous cell carcinoma. Clin. Exp. Dent. Res. 2018, 4, 255–262.
- Mukherjee, P.K.; Chandra, J.; Retuerto, M.; Sikaroodi, M.; Brown, R.E.; Jurevic, R.; Salata, R.A.; Lederman, M.M.; Gillevet, P.M.; Ghannoum, M.A. Oral mycobiome analysis of HIV-infected patients: Identification of Pichia as an antagonist of opportunistic fungi. PLoS Pathog. 2014, 10, e1003996.
- Zakaria, M.N.; Furuta, M.; Takeshita, T.; Shibata, Y.; Sundari, R.; Eshima, N.; Ninomiya, T.; Yamashita, Y. Oral mycobiome in community-dwelling elderly and its relation to oral and general health conditions. Oral Dis. 2017, 23, 973–982.
- Ahmed, N.; Ghannoum, M.; Gallogly, M.; de Lima, M.; Malek, E. Influence of gut microbiome on multiple myeloma: Friend or foe? J. Immunother. Cancer 2020, 8.
- Chung, L.M.; Liang, J.A.; Lin, C.L.; Sun, L.M.; Kao, C.H. Cancer risk in patients with candidiasis: A nationwide population-based cohort study. Oncotarget 2017, 8, 63562–63573.
- Chimonidou, M.; Strati, A.; Tzitzira, A.; Sotiropoulou, G.; Malamos, N.; Georgoulias, V.; Lianidou, E.S. DNA methylation of tumor suppressor and metastasis suppressor genes in circulating tumor cells. Clin. Chem. 2011, 57, 1169–1177.
- Enroth, H.; Kraaz, W.; Engstrand, L.; Nyrén, O.; Rohan, T. Helicobacter pylori strain types and risk of gastric cancer: A case-control study. Cancer Epidemiol. Biomark. Prev. 2000, 9, 981–985.
- Blaser, M.J.; Perez-Perez, G.I.; Kleanthous, H.; Cover, T.L.; Peek, R.M.; Chyou, P.H.; Stemmermann, G.N.; Nomura, A. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995, 55, 2111–2115.
- Peters, B.A.; Wu, J.; Hayes, R.B.; Ahn, J. The oral fungal mycobiome: Characteristics and relation to periodontitis in a pilot study. BMC Microbiol. 2017, 17, 157.
- Sung, K.H.; Josewski, J.; Dübel, S.; Blankenfeldt, W.; Rau, U. Structural insights into antigen recognition of an anti-β-(1,6)-β-(1,3)-D-glucan antibody. Sci. Rep. 2018, 8, 13652.
- Kimura, Y.; Tojima, H.; Fukase, S.; Takeda, K. Clinical evaluation of sizofilan as assistant immunotherapy in treatment of head and neck cancer. Acta Otolaryngol. Suppl. 1994, 511, 192–195.
- Mansour, A.; Daba, A.; Baddour, N.; El-Saadani, M.; Aleem, E. Schizophyllan inhibits the development of mammary and hepatic carcinomas induced by 7,12 dimethylbenz(α)anthracene and decreases cell proliferation: Comparison with tamoxifen. J. Cancer Res. Clin. Oncol. 2012, 138, 1579–1596.
- Okamura, K.; Suzuki, M.; Chihara, T.; Fujiwara, A.; Fukuda, T.; Goto, S.; Ichinohe, K.; Jimi, S.; Kasamatsu, T.; Kawai, N.; et al. Clinical evaluation of schizophyllan combined with irradiation in patients with cervical cancer. A randomized controlled study. Cancer 1986, 58, 865–872.
- Luan, C.; Xie, L.; Yang, X.; Miao, H.; Lv, N.; Zhang, R.; Xiao, X.; Hu, Y.; Liu, Y.; Wu, N.; et al. Dysbiosis of fungal microbiota in the intestinal mucosa of patients with colorectal adenomas. Sci. Rep. 2015, 5, 7980.
- Gao, R.; Kong, C.; Li, H.; Huang, L.; Qu, X.; Qin, N.; Qin, H. Dysbiosis signature of mycobiota in colon polyp and colorectal cancer. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 2457–2468.
- Richard, M.L.; Liguori, G.; Lamas, B.; Brandi, G.; da Costa, G.; Hoffmann, T.W.; Pierluigi Di Simone, M.; Calabrese, C.; Poggioli, G.; Langella, P.; et al. Mucosa-associated microbiota dysbiosis in colitis associated cancer. Gut Microbes 2018, 9, 131–142.
- Coker, O.O.; Nakatsu, G.; Dai, R.Z.; Wu, W.K.K.; Wong, S.H.; Ng, S.C.; Chan, F.K.L.; Sung, J.J.Y.; Yu, J. Enteric fungal microbiota dysbiosis and ecological alterations in colorectal cancer. Gut 2019, 68, 654–662.
- Aykut, B.; Pushalkar, S.; Chen, R.; Li, Q.; Abengozar, R.; Kim, J.I.; Shadaloey, S.A.; Wu, D.; Preiss, P.; Verma, N.; et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 2019, 574, 264–267.
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732.
- Zhang, J.; Haines, C.; Watson, A.J.M.; Hart, A.R.; Platt, M.J.; Pardoll, D.M.; Cosgrove, S.E.; Gebo, K.A.; Sears, C.L. Oral antibiotic use and risk of colorectal cancer in the United Kingdom, 1989-2012: A matched case-control study. Gut 2019, 68, 1971–1978.
- Qin, X.; Gu, Y.; Liu, T.; Wang, C.; Zhong, W.; Wang, B.; Cao, H. Gut mycobiome: A promising target for colorectal cancer. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188489.
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Hoffmann, C.; Dollive, S.; Grunberg, S.; Chen, J.; Li, H.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Archaea and fungi of the human gut microbiome: Correlations with diet and bacterial residents. PLoS ONE 2013, 8, e66019.
- Dai, Z.; Zhang, J.; Wu, Q.; Chen, J.; Liu, J.; Wang, L.; Chen, C.; Xu, J.; Zhang, H.; Shi, C.; et al. The role of microbiota in the development of colorectal cancer. Int. J. Cancer 2019, 145, 2032–2041.
- Song, M.; Chan, A.T.; Sun, J. Influence of the Gut Microbiome, Diet, and Environment on Risk of Colorectal Cancer. Gastroenterology 2020, 158, 322–340.
- Trojanowska, D.; Zwolinska-Wcislo, M.; Tokarczyk, M.; Kosowski, K.; Mach, T.; Budak, A. The role of Candida in inflammatory bowel disease. Estimation of transmission of C. albicans fungi in gastrointestinal tract based on genetic affinity between strains. Med. Sci. Monit. 2010, 16, Cr451–Cr457.
- Liguori, G.; Lamas, B.; Richard, M.L.; Brandi, G.; da Costa, G.; Hoffmann, T.W.; Di Simone, M.P.; Calabrese, C.; Poggioli, G.; Langella, P.; et al. Fungal Dysbiosis in Mucosa-associated Microbiota of Crohn’s Disease Patients. J. Crohns Colitis 2016, 10, 296–305.
- Li, J.; Chen, D.; Yu, B.; He, J.; Zheng, P.; Mao, X.; Yu, J.; Luo, J.; Tian, G.; Huang, Z.; et al. Fungi in Gastrointestinal Tracts of Human and Mice: From Community to Functions. Microb. Ecol. 2018, 75, 821–829.
- Qiu, X.; Zhang, F.; Yang, X.; Wu, N.; Jiang, W.; Li, X.; Li, X.; Liu, Y. Changes in the composition of intestinal fungi and their role in mice with dextran sulfate sodium-induced colitis. Sci. Rep. 2015, 5, 10416.
- Mueller, K.D.; Zhang, H.; Serrano, C.R.; Billmyre, R.B.; Huh, E.Y.; Wiemann, P.; Keller, N.P.; Wang, Y.; Heitman, J.; Lee, S.C. Gastrointestinal microbiota alteration induced by Mucor circinelloides in a murine model. J. Microbiol. 2019, 57, 509–520.
- Mason, K.L.; Erb Downward, J.R.; Mason, K.D.; Falkowski, N.R.; Eaton, K.A.; Kao, J.Y.; Young, V.B.; Huffnagle, G.B. Candida albicans and bacterial microbiota interactions in the cecum during recolonization following broad-spectrum antibiotic therapy. Infect. Immun. 2012, 80, 3371–3380.
- Vallianou, N.; Dalamaga, M.; Stratigou, T.; Karampela, I.; Tsigalou, C. Do Antibiotics Cause Obesity Through Long-term Alterations in the Gut Microbiome? A Review of Current Evidence. Curr. Obes. Rep. 2021, 1–19.
- Santus, W.; Devlin, J.R.; Behnsen, J. Crossing Kingdoms: How the Mycobiota and Fungal-Bacterial Interactions Impact Host Health and Disease. Infect. Immun. 2021, 89.
- Yang, W.; Zhou, Y.; Wu, C.; Tang, J. Enterohemorrhagic Escherichia coli promotes the invasion and tissue damage of enterocytes infected with Candida albicans in vitro. Sci. Rep. 2016, 6, 37485.
- Lambooij, J.M.; Hoogenkamp, M.A.; Brandt, B.W.; Janus, M.M.; Krom, B.P. Fungal mitochondrial oxygen consumption induces the growth of strict anaerobic bacteria. Fungal Genet. Biol. 2017, 109, 1–6.
- Sánchez-Alonzo, K.; Parra-Sepúlveda, C.; Vega, S.; Bernasconi, H.; Campos, V.L.; Smith, C.T.; Sáez, K.; García-Cancino, A. In Vitro Incorporation of Helicobacter pylori into Candida albicans Caused by Acidic pH Stress. Pathogens 2020, 9, 489.
- Van Leeuwen, P.T.; van der Peet, J.M.; Bikker, F.J.; Hoogenkamp, M.A.; Oliveira Paiva, A.M.; Kostidis, S.; Mayboroda, O.A.; Smits, W.K.; Krom, B.P. Interspecies Interactions between Clostridium difficile and Candida albicans. mSphere 2016, 1.
- Tomkovich, S.; Dejea, C.M.; Winglee, K.; Drewes, J.L.; Chung, L.; Housseau, F.; Pope, J.L.; Gauthier, J.; Sun, X.; Mühlbauer, M.; et al. Human colon mucosal biofilms from healthy or colon cancer hosts are carcinogenic. J. Clin. Investig. 2019, 129, 1699–1712.
- Hager, C.L.; Ghannoum, M.A. The mycobiome: Role in health and disease, and as a potential probiotic target in gastrointestinal disease. Dig. Liver Dis. 2017, 49, 1171–1176.
- Wu, J.; Li, Q.; Fu, X. Fusobacterium nucleatum Contributes to the Carcinogenesis of Colorectal Cancer by Inducing Inflammation and Suppressing Host Immunity. Transl. Oncol. 2019, 12, 846–851.
- Spyrou, N.; Vallianou, N.; Kadillari, J.; Dalamaga, M. The interplay of obesity, gut microbiome and diet in the immune check point inhibitors therapy era. Semin. Cancer Biol. 2021, 73, 356–376.
- Karpiński, T.M. Role of Oral Microbiota in Cancer Development. Microorganisms 2019, 7, 20.
- Sinha, R.; Ahn, J.; Sampson, J.N.; Shi, J.; Yu, G.; Xiong, X.; Hayes, R.B.; Goedert, J.J. Fecal Microbiota, Fecal Metabolome, and Colorectal Cancer Interrelations. PLoS ONE 2016, 11, e0152126.
- Wu, T.; Cen, L.; Kaplan, C.; Zhou, X.; Lux, R.; Shi, W.; He, X. Cellular Components Mediating Coadherence of Candida albicans and Fusobacterium nucleatum. J. Dent. Res. 2015, 94, 1432–1438.
- Marzano, M.; Fosso, B.; Piancone, E.; Defazio, G.; Pesole, G.; De Robertis, M. Stem Cell Impairment at the Host-Microbiota Interface in Colorectal Cancer. Cancers 2021, 13, 996.
- Arnold, M.; Abnet, C.C.; Neale, R.E.; Vignat, J.; Giovannucci, E.L.; McGlynn, K.A.; Bray, F. Global Burden of 5 Major Types of Gastrointestinal Cancer. Gastroenterology 2020, 159, 335–349.
- Dalamaga, M.; Polyzos, S.A.; Karmaniolas, K.; Chamberland, J.; Lekka, A.; Migdalis, I.; Papadavid, E.; Dionyssiou-Asteriou, A.; Mantzoros, C.S. Circulating fetuin-A in patients with pancreatic cancer: A hospital-based case-control study. Biomarkers 2014, 19, 660–666.
- Dalamaga, M.; Migdalis, I.; Fargnoli, J.L.; Papadavid, E.; Bloom, E.; Mitsiades, N.; Karmaniolas, K.; Pelecanos, N.; Tseleni-Balafouta, S.; Dionyssiou-Asteriou, A.; et al. Pancreatic cancer expresses adiponectin receptors and is associated with hypoleptinemia and hyperadiponectinemia: A case-control study. Cancer Causes Control. 2009, 20, 625–633.
- Sam, Q.H.; Chang, M.W.; Chai, L.Y. The Fungal Mycobiome and Its Interaction with Gut Bacteria in the Host. Int. J. Mol. Sci. 2017, 18, 330.

