1. Introduction
Fungi have recently been estimated to consist of up to 3.8 million species; thus, they represent a taxonomic and functional diversity of life forms, being implicated in complex and yet unknown interactions with other living microorganisms
[1]. Fungi are microeukaryotes and constitute a smaller part of the human microbiome in comparison to bacteria, forming the so-called “human mycobiome”
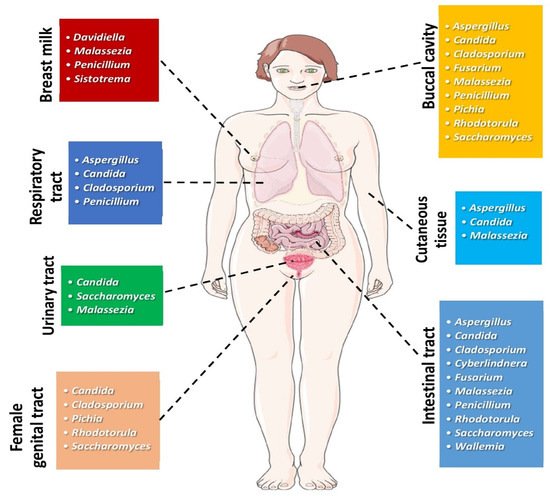
[2][3][4][2,3,4]. Fungal communities can be found in different anatomic sites of the human body, as depicted in
Figure 1.
Figure 1. Most common genera of fungi found in different human body sites under physiologic conditions. (All images originate from the free medical website
http://smart.servier.com/ (accessed on 25 May 2021) by Servier licensed under a Creative Commons Attribution 3.0 Unported License).
The number of human microbiota has been determined to be 10
14, about 10 times greater than the number of human cells. Also, the quantity of microbial genes is about 100 times more than the corresponding quantity of human genes. The human mycobiome accounts for approximately 0.001% to 0.1% of the microbial community in the gut
[5][6][5,6]. Over the last years, fungi have been the subject of intense investigation, with a particular focus on their contribution to human disorders, especially among immune-compromised patients
[7]. However, as most fungi are not easily cultured, even in specific cultural media, their study has been limited until today, due mainly to the unavailability of methods used for their detection. Nevertheless, genomic methodology in fungi research may broaden our knowledge in their contribution to health and disease
[2][3][4][2,3,4]. High-throughput sequencing (HTS) analysis of fungi is reshaping the area of the fungal community
[1].
In the gut, bacteria outnumber fungi, but we cannot overlook the fact that fungal taxa may merely be determined with modern sophisticated, non-culture-based methods. Despite the fact that the gut mycobiome is less analyzed than the bacteriome, it seems likely that fungi are primarily spread intra-uterinally to the fetus
[8]. Recent studies have suggested that fungi are found in the gut microflora of young children via transmission from their mother, siblings and environmental exposure; nevertheless, diet may be the most significant factor
[9]. Dietary intake plays a key role, as fungi colonize the gut by food digestion. Fungi which colonize the intestines via dietary intake could be part of the gut flora or be rejected
[3]. Despite the paucity of studies, the importance of dietary intake in the content of the intestinal mycobiome is confirmed by the fact that vegetarians present dissimilarities in the mycobiotic composition in comparison to those following a Western-style nutrition
[3][10][3,10]. The interplay of intestinal mycobiome with bacteriome and virome is a hot topic of research, especially in the field of mycobiome-associated diseases.
Current scientific evidence has supported the contribution of the intestinal mycobiome in affecting immune response, with an impact on regional and systemic disease
[11]. Notably, a considerable number of pathogenic fungi are “pathobionts”, i.e., residents in the organism that are not implicated in the pathogenesis of any disorders under physiologic circumstances but that may exhibit pathogenetic properties. Following this trend,
Candida albicans, which belongs to the physiologic intestinal ecosystem, is the etiologic agent of systemic candidiasis in immune-compromised subjects
[12]. The transformation of non-pathogenic fungi under physiologic circumstances to pathogenic fungi under unspecified conditions is a subject of intense research. Indeed, fungal diseases constitute a considerable part of the totality of the infectious disease range. A substantial part of infections includes fungal infections in immune-compromised subjects with an approximate death rate of 35% to 45%
[12]. However, there is currently growing interest in the associations between the human mycobiome and its potential role in human carcinogenesis. In this comprehensive review, we present a synopsis of recent data on the human mycobiome and cancer, focusing on specific cancer types based on current available scientific evidence, giving an emphasis on the interplay among the human mycobiome, microbiome and the host influencing carcinogenesis.
2. Mycobiome and Head and Neck Cancer
Head and neck cancer is the 6th most frequent malignancy globally, with oral cancer (OC) and oropharyngeal carcinoma (OPC) being the most common types. Approximately half of the cases of OC and OPC have topical or remote metastases at diagnosis, thus resulting in a 50% death rate
[13][14][13,14]. The risk factors of head and neck squamous cell carcinoma (HNSCC) have not been elucidated until today. Main etiologic factors of HNSCC include human papilloma virus (HPV), tobacco, genetic predisposition, UV radiation, alcohol consumption, occupational exposure to wood and coal dust, asbestos, formaldehyde, and nutrition poor in vegetables and fruits
[14][15][14,15].
The role of the mycobiome in OC and OPC has not been thoroughly investigated.
Candida spp. are the most commonly encountered fungi in the oral mycobiome among healthy adults, followed by
Cladosporium,
Aureobasidium,
Saccharomycetales,
Aspergillus,
Fusarium and
Cryptococcus. In particular,
Candida,
Aspergillus,
Fusarium and
Cryptococcus represent the leading genera, and are considered pathogenic fungi in humans
[16]. Nevertheless, there is a paucity of data regarding the oral fungal community amid patients with cancer. Shelburne et al. have studied host whole exome sequencing as well as genetic analysis of infectious agents, and have determined the oral and fecal microbiome and mycobiome in a patient with leukemia. They concluded that bacterial dysbiosis in the oral cavity could provide a permissive milieu for the subsequent emergence of invasive mucormycosis
[17]. Furthermore, recent studies have highlighted the importance of the interplay between bacterial and fungal communities, i.e., inter-kingdom interplay. These studies have pointed out that the bacteriome or the mycobiome could contribute to the pathogenesis of various diseases, but their interaction may also have an important impact
[17]. In order to examine the interaction between oropharyngeal bacteriome and the mycobiome, Mukheerjie et al. have focused on random forest modeling of an oral mycobiome and bacteriome
[18]. Amid the predominant parameters, this model has detected ten genera of bacteria, such as
Rothia,
Eikenella,
Streptococcus,
Porphyromonas,
Aggregatibacter,
Fusobacterium,
Prevotella,
Actinomyces,
Campylobacter,
Capnocytophaga, and only one genus of fungi,
Emericella, Afterwards, they performed inter-and intra-kingdom association analyses with taxa belonging to bacteria and fungi in the microbiota of 39 oral tongue cancer and non-tumor samples. They have demonstrated that
Bacteroidetes showed positive intra-kingdom associations with
Fusobacteria and
Spirochaetes in cancer samples. In parallel, there was a negative relationship between
Zygomycota and
Ascomycota, whilst the association between
Glomeromycota and
Ascomycota was reduced in cancer samples. In addition,
Zygomycota had a positive inter-kingdom relationship with
Fusobacteria and
Bacteroidetes, and a negative relationship with
Actinobacteria. Fungal species such as
Lichtheimia presented a positive association with
Campylobacter,
Porphyromonas and
Fusobacterium, and a negative one with
Actinomyces. Lichtheimia corymbifera, a member of the
Mucoraceae family in the
Zygomycota phylum, was found to be positively related to eleven bacteria and negatively to thirty-nine bacteria, among which was
Lactobacillus spp. These findings shed light on the specific inter- and intra-kingdom relationship that may take place in the bacterial and fungal communities in the context of oral tongue cancer
[18].
Given the complexity of carcinogenesis, we may hypothesize that many genomic and epigenomic loci exhibit alterations in head and neck malignant neoplasms, confirming the multi-hit process of malignancy. Mukheerjie et al. have documented a similar multi-hit process of bacteriome and mycobiome in the etiopathogenesis of oral carcinogenesis. Alterations in the oral microbiome and mycobiome may account for cancerous effects of metabolites secreted by these microorganisms. In this context, acetaldehyde, which is produced by alcohol metabolism, was suggested to be linked to OC related to chronic alcohol intake. Due to chronic alcohol consumption and abundance of bacteria which synthesize acetaldehyde, including
Rothia,
Streptococcus and
Prevotella, higher oral acetaldehyde may be implicated in oral tumorigenesis
[18]. Cancer and non-cancer groups presented differences in fungal abundance. Some fungi could exhibit an oncogenic potential, as shown with
Candida albicans, which may participate in the synthesis of salivary acetaldehyde in subjects with ethanol-associated OC
[19][20][21][22][23][19,20,21,22,23]. More research is needed to also explore the carcinogenic properties of the fungi
Lichtheimia corymbifera. It is noteworthy that correlation analyses have also documented a negative association between
Lichtheimia corymbifera and
Lactobacillus spp., that may be associated with alterations in the regional intestinal milieu that enhances the overgrowth of particular taxa.
Lactobacillus spp. are considered favorable bacteria that modulate the development of bacterial and fungal communities
[3][23][3,23]. A decrease of
Lactobacillus spp. could cause perturbations in the microbial microflora of patients suffering from oral tongue cancer. This imbalance in the microbial ecosystem may interfere with factors, such as pH, and/or micronutrients, which predispose to microbial dysbiosis
[23].
Very recently, Shay et al. have studied the bacterial and fungal communities as well as their interplay in the oral wash of forty-six subjects with HNSCC and a similar number of non-HNSCC individuals
[24]. Oral wash samples were collected for microbiome studies. They have detected three phyla of fungi and eleven phyla of bacteria.
Ascomycota from the fungal community (72%) and
Firmicutes from the bacterial community (39%) were the predominant microorganisms. Notably, strains of
Candida albicans and
Rothia mucilaginosa presented differences in abundance, whereas
Schizophyllum commune was diminished in the oral wash from subjects suffering from HNSCC in comparison to non-HNSCC individuals. Collectively, these findings highlight the existence of differences in abundance of bacterial and fungal communities as well as the microbiome–mycobiome interactions in the oral wash of subjects with HNSCC, in comparison to non-HNSCC participants. In particular, specific strains of
Candida albicans were over-presented or under-presented in the oral wash samples from subjects with malignancies, when compared to samples from non-HNSCC participants.
Candida dubliniensis,
Schizophyllum commune and a fungus from the class of
Agaricomycetes were over-represented in controls in comparison to cancer patients. On the contrary, one fungal strain of
Neoascochyta exitialis was under-represented in the oral wash from subjects with HNSCC, in comparison to controls.
Candida was the predominant fungal genus in the oral fungal microflora of both patients with HNSCC and non-HNSCC participants
[24]. This finding has been observed across many studies examining the oral mycobiome among patients and controls
[25][26][27][25,26,27].
Oral candidiasis has been related to the development of malignancies, such as head and neck malignancies
[25][26][27][25,26,27]. Perera et al. have detected an overgrowth of
Candida albicans in the oral squamous cell malignant tissue in comparison to benign tissue (intra-oral fibro-epithelial polyps)
[26]. Vesty et al. have noted an enrichment of
Candida albicans in the saliva of subjects with HNSCC patients, which correlated with an increase in the inflammatory cytokines interleukin (ΙL)-1β and IL-8
[27]. The latter observation is suggestive of the potential contribution of
Candida albicans to the promotion of inflammation and carcinogenesis through hyper-methylation of various tumor suppressor genes
[28][29][28,29]. In addition,
Candida albicans is known to produce biofilms, which form a resistant shield that protects the fungal community from external factors, and are related to improper immune elimination by the host. Fungal filamentation is also a known
Candida virulence factor, which also damages host tissues and triggers host inflammatory response
[30]. Nevertheless, abundance of
C. albicans in both healthy participants and patients does not provide enough evidence that this organism may be implicated in HNSCC carcinogenesis
[30][31][32][30,31,32]. It is plausible that the study by Shay et al. identified both pathogenic and non-pathogenic
C. albicans strains. Further research is necessary to characterize those
C. albicans strains that are related to HNSCC
[24]. This characterization could increase the specificity of a microbiome-based oral wash screening tool for HNSCC. Apart from the differential species of
C. albicans, a second fungi,
Schizophyllum commune, was in abundance in the oral wash of healthy controls. The genera
Schizophyllum is a member of the phylum
Basidiomycota, and has been known as a member of the oral mycobiome
[33][34][35][33,34,35].
Schizophyllum commune is suggested to produce the polysaccharide compound schizophylan
[36]. Schizophylan has anti-cancerous properties in vitro and has shown promise in the treatment of cancer patients, including HNSCC, in studies conducted in Japan in the 1980s
[35][36][37][38][39][35,36,37,38,39]. The abundance of
Schizophyllum commune among controls supports its role as a potential anticancer agent.
Table 1 depicts the main studies associating the mycobiome with neoplastic diseases in animal models and in humans.
Table 1. List of main studies associating the mycobiome with various types of neoplasms in animal models and humans.
| Research/Year |
Population, Type of Study |
Clinical Specimen |
Main Findings |
Remarks |
| Head and Neck Cancer |
| Perera et al., 2017 [26] |
52 individuals; 25 with OSCC; 27 intra-oral-fibro epithelial polyps |
52 biopsies from 25 patients with OSCC and 27 with oral polyps. DNA was extracted and sequenced for the ITS2 region |
364 species accounting for 160 genera and 2 phyla ( | Ascomycota | and | Basidiomycota | ) were detected.
| Candida | and | Malassezia | made up 48% and 11% of the average fungal community, respectively, according to Luan et al., 2015. |
-5 species and 4 genera were identified in more than half of samples.
-Less abundance and diversity in OSCC tissues of patients.
| -Candida | , | Hannaella | , and | Gibberella | were ↑↑ in OSCC; | Altenaria | and | Trametes | were in greater quantity in polyps specimens.
- | Candida albicans | , | Candida etchellsii | , and | Hannaella luteola | –like species were enriched in OSCC | Hanseniaspora uvarum | –like species, | Malassezia restricta | , and | Aspergillus tamarii | are predominant in polyps specimens.
-Dysbiotic mycobiome dominated by | C. albicans | has been observed in OSCC. |
| Mukherjee et al., 2017 [25] |
39 participants with OSCC of the tongue |
39 tissue samples from oral SCC and adjacent tissues were analyzed after DNA extraction for 16S/18S rRNA gene. |
Fungal richness was ↓↓ in tumor tissue (TT) in comparison to the adjacent non-cancerous tissue (ANCT), | p | < 0.006.
The presence of 22 bacterial and 7 fungal genera was different in TT and ANCT.
| Aspergillus | in TT was negatively associated with the presence of bacteria | Actinomyces | , | Prevotella | , | Streptococcus | , whilst it presented a positive association with | Aggregatibacter | . |
-Subjects with advanced T-stage disease presented reduced mean differences between TT and ANCT, in comparison to subjects with regional disease.
-Findings indicative of differences in the bacteriome and mycobiome between OSCC patients and their adjacent non-cancerous oral epithelium
-Association with T-stage.
-Despite the similarities in the index of diversity of the mycobiome between TT and ANCT, the abundance of the mycobiome was diminished in TT.
-This study is suggestive of existing changes in the local environment in patients with OSCC, expressed as specific bacterial and fungal dysbiosis |
| Vesty et al., 2018 [27] |
30 participants, including 14 patients with HNSCC |
Saliva specimens analyzed by 16S rRNA gene and ITS1amplicon sequencing |
↑↑ | Candida |
| Candida albicans | representing more than 96% of fungi in the majority of subjects with HNSCC. |
-↑↑ IL-1β and IL-8 in HNSCC and patients with poor dental health, when compared to healthy controls.
-IL-1β and IL-8 levels were associated with | C. albicans | .
-In HNSCC, salivary microbial and inflammatory markers are affected by oral hygiene. |
| Shay et al., 2020 [24] |
92 individuals, including 46 patients with HNSCC |
Oral wash samples analyzed by 16S rRNA and ITS gene sequencing |
Distinct strains of | Candida albicans | are increased or decreased in oral wash specimens from patients with HNSCC, when compared to healthy controls. |
-Distinct strains of | Candida albicans | and | Rothia mucilaginosa | differed in numbers. | Schizophyllum | commune was decreased in HNSCC patients, in comparison to healthy controls.
-Compared to controls, oral cavity of subjects with HNSCC presents distinct differences in the mycobiome and bacteriome, and their interactions. |
| Colorectal Cancer |
| Luan et al., 2015 [40] |
27 patients with colorectal adenomas |
Biopsies from colorectal adenomas and adjacent tissues were studied by using denaturing gradient gel electrophoresis (DGGE) |
↑↑ | Ascomycota | , | Glomeromycota | and | Basidiomycota | .
↓↓ diversity in adenomas compared to adjacent tissue |
-↑↑ | Basidiomycota | in adjacent tissues.
-↑↑ | Basidiomycota | and | Saccharomycetales | in advanced adenoma samples, when compared to non-advanced. |
| Gao et al., 2017 [41] |
131 individuals with colorectal carcinoma (CRC), colorectal polyps and normal subjects |
Stool samples from patients with CRC, polyps and normal subjects were analyzed by using ITS2 gene sequencing |
↑ ↑ | Ascomycota | followed by | Basidiomycota |
↓↓ diversity in the polyp group, when compared to controls. |
↑↑ Ratio of | Ascomycota | to | Basidiomycota | in subjects with CRC and polyps, in comparison to controls.
↑↑ of the opportunistic fungi | Trichosporon | and | Malassezia | , which could be implicated in the progression to CRC. |
| Richard et al., 2018 [42] |
27 patients with CRC; 7 with colitis-associated cancer, 10 patients with sporadic cancer and 10 healthy individuals |
Tissue specimens from colonic resections in colitis-associated malignancy and sporadic CRC groups were analyzed using 16S rRNA and ITS2 sequencing |
↑↑ | Basidiomycota | followed by | Ascomycota |
↓ diversity in sporadic cancer. |
↑↑ | Basidiomycota | in colitis-associated cancer. |
| Coker et al., 2019 [43] |
585 individuals; 184 patients with CRC, 197 patients colorectal adenomas and 204 normal subjects |
Stool samples from patients with CRC, colorectal adenomas and normal subjects were analyzed by fecal shotgun metagenomic sequencing |
- | Ascomycota | , | Basidiomycota | and | Mucoromycota | in patients with CRC and healthy participants.
-No difference in diversity |
-↑↑ | Basidiomycota/Ascomycota | ratio in CRC when compared to controls.
-14 fungi identified with differential composition between CRC and controls. |
| Pancreatic Cancer |
| Aykut et al., 2019 [44] |
(1) Experiments in mice as well as in humans using 18S rRNA sequencing
KC mice, which develop spontaneous pancreatic cancer by targeted expression of mutant Kras. C57BL/6, MBL-null, and C3−/− mice.
(2) Human stool samples and pancreatic tissue specimens were gathered from healthy volunteers and subjects undergoing surgery for PDA or benign pancreatic disorder. |
Because of the direct proximity and relationship of the intestinal and pancreatic duct via the Oddi sphincter, gut fungi could enter the pancreas. To examine this hypothesis, they administered GFP-labeled | Saccharomyces cerevisiae | to controls or cancer-bearing mice through oral gavage. Fungi moved into the pancreas in less than thirty minutes, suggesting that the intestinal fungal community may directly impact on the pancreatic microenvironment. |
-PDA tumors harbored a ~3000-fold augmentation in fungi, in comparison to physiologic pancreas in both mice and humans.
-PDA mycobiome was different from gut or physiologic pancreatic mycobiome based on diversity indexes.
-The fungal community infiltrating PDA was ↑↑ enriched in | Malassezia | in mice and humans.
-Fungal elimination with the use of amphotericin B was tumor-protective in slowly progressive as well as in models of invasive PDA, whereas re-population with | Malassezia | but not | Candida | , | Saccharomyces | , or | Aspergillus | –promoted oncogenesis. |
-Connection of mannose-binding lectin (MBL), that attaches fungal wall glycans to activate the complement pathway, was needed in the promotion of malignancy.
-MBL or C3 deletion in the extra-tumoral area or C3aR knockdown in tumor cells prevented tumor expansion. Reprogramming of the fungal ecosystem did not change PDA progression in MBL or C3 deficient mice.
-Pathogenic fungi may promote PDA by activating the complement pathway via MBL induction. |
Overall, while some C. albicans strains are involved in the etiopathogenesis of HNSCC, other strains are not participating. Moreover, Schizophyllum commune seems to be protective against HNSCC. It remains to be elucidated whether it is just the specific strains or the inter-kingdom interplay with large-scale, longitudinal, multi-omics studies combining metagenomics and metabolomics.