 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Josko Bozic | + 4276 word(s) | 4276 | 2021-06-21 08:26:46 | | | |
| 2 | Conner Chen | Meta information modification | 4276 | 2021-06-28 09:45:14 | | | | |
| 3 | Conner Chen | Meta information modification | 4276 | 2021-06-30 10:31:35 | | |
Video Upload Options
Causing more than 1.8 million deaths a year worldwide, acute coronary syndrome (ACS) represents a major global health burden. It has so far been well-established that most of the plaques leading to ACS are not a result of gradual narrowing of the vessel lumen, but rather a result of sudden disruption of vulnerable atherosclerotic plaques. Apart from various imaging modalities, multiple biomarkers were proposed to identify the presence of vulnerable plaques or plaques at different stages of remodeling. A logic behind measurement of biomarkers in blood that reflect the presence of vulnerable plaque is dual. Certain molecules could arise as a consequence of leakage from unstable plaques, whereas others could merely indicate that a patient is susceptible to the development of a vulnerable plaque as those molecules are implicated in plaque destabilization.
1. Pathophysiology of Vulnerable Plaques
The detrimental cascade by which atherosclerosis develops in arteries is one of the most studied processes in humans [1]. However, not all of the aspects have been completely understood so far [2]. Changes in the constitutive properties of endothelial cells (ECs) on arterial-susceptible sites with increased endothelial shear stress (branch points and the outer wall of bifurcations) are the initial step of atherosclerotic plaque development [3]. Under the effects of subtle microenvironmental triggers from blood or interstitia, vascular ECs become dysfunctional and begin to synthesize an array of cytokines and chemokines whilst upregulating the expression of various cell-adhesion molecules, setting off a complex cascade that results in atherosclerotic plaque formation [4][5][6][7][8][9][10].
The crucial stage of the atherosclerotic process is creation of the unstable fibro-lipid plaque. However, it is now very important to acknowledge that, for reasons not completely understood, the vast majority of the atherosclerotic lesions, as many as 95%, will never result in an acute thrombo-occlusive vascular disease or any symptoms for that matter [11][12][13]. Recent analyses suggest that those vulnerable, rupture-prone plaques fall into two categories: The first being plaques with lipid-rich necrotic cores, thin fibrous caps and abundant in inflammatory cells, and the other being plaques characterized by excessive extracellular matrix and endothelial apoptosis [14]. Plaques with necrotic cores are primarily associated with ST-elevation myocardial infarction (STEMI) and their pathobiology has been well understood. The necrotic core of those plaques is a result of death of macrophages but also smooth muscle cells (SMCs) in the lesion combined with impairment in efferocytosis—i.e., poor phagocytic clearance of dead cells. The rupture itself is a consequence of fibrous cap thinning which arises from death of collagen-producing SMCs in the intima and upregulation of matrix-destroying proteases, whereas lipid-loaded core creates additional physical strain on the overlying fibrous cap making it even more susceptible to the rupture [15].
Unlike the former, matrix-rich lipid-poor plaques usually lack prominent macrophage collections and the main pathophysiologic mechanism that leads to ACS in these circumstances is superficial erosion, a poorly understood process not pertaining to pathogenetic mechanisms associated with plaque rupture [16][17][18][19]. As discussed by Libby et al., it seems that multiple processes predispose these plaques to superficial erosion, including flow disturbance, basement membrane breakdown, endothelial cell death, and detachment potentiated by innate immune activation mediated through pattern-recognition receptors, as well as endothelial–mesenchymal transition [20]. It is suggested that at least 50% of ACS arise as a consequence of plaque rupture, around 33% as a result of plaque erosion, whereas for the rest of ACS we have no clear proofs of either rupture or erosion [21][22][23][24].
Although currently there is no definitive evidence, it seems that the principal contributor to the plaque progression from virtually harmless stable into vulnerable rupture-prone plaque is an interplay between the aforementioned impaired efferocytosis and defective resolution of the inflammation in the advanced stages of atherosclerosis. The mechanism of defective efferocytosis in plaque progression is still largely misunderstood, yet it is quite clear that overwhelming apoptosis is not a contributor to the process [25]. Multiple authors favor the hypothesis that specific molecular-cellular processes involved in uptake or recognition of apoptotic cells by intralesional macrophages lead to impaired efferocytosis [26][27].
The concept of defective resolution implies failure of mechanisms that usually lead to repair of the inflammation-affected areas [28]. The resolution of inflammation is actually a very primal defense mechanism, that evolved concomitantly with inflammation in order to restore tissue function which has been deteriorated by inflammation’s detrimental effects. The resolution is mediated by multiple effector arms [29]: endogenous lipids called specialized pro-resolving mediators (SPMs), which include lipoxins, resolvins, protectins, and maresins; certain bioactive gases (nitric oxide, hydrogen sulphide, and carbon monoxide); anti-inflammatory proteins (IL-10, TGFβ, and annexin A); and resolving cells which include regulatory T cells (Treg) and resolving-type macrophages. In states of chronic inflammation, such as atherosclerosis, these effectors are impaired and lead to an amplification cycle of continuous tissue injury [30]. Multiple evidence suggests that atherosclerotic plaques have reduced levels of the above-noted effectors of resolution [30][31]. Accordingly, exogenous administration of these molecules has been shown to suppress the progression of mid-stage plaques to advanced plaques [32][33][34].
However, it still remains unclear as to what dampens the resolution in the setting of atherosclerotic plaque progression. There are three possible targets within the resolution framework that could be targeted: the production of resolution agents, excessive inactivation of those agents and finally, impaired response to those agents [35]. Multiple studies suggest that all of those mechanisms take part in impairment of resolution [32][33][36]. Interestingly, studies suggest that in phagocytes which accumulate apoptotic cells, i.e., when efferocytosis is preserved, production of SPMs is upregulated [37]. In addition, activation of the MerTK receptor, macrophage receptor involved in the process of efferocytosis, results in upregulation of SPMs production as well [38]. These observations highlight the mutual relationship between efferocytosis and resolution, two of the most viable mechanisms involved in progression of vulnerable atherosclerotic plaques. Finally, it is important to note that following the CV event, atherosclerotic plaque stabilizes over time, as demonstrated by Peeters et al. [39]. Plaque stabilization can be achieved by various mechanisms: by increasing of fibrous cap thickness, by reduction of inflammation in the fibrous cap and by reducing size of atheromatous core [40]. It is also noteworthy that plaques may be stabilized against thrombosis independent of changes in either plaque size or luminal obstruction [40].
2. Biomarkers of Vulnerable Plaques
Since atherosclerosis is an everlasting field of research worldwide, there is a multitude of biomarkers whose potential to indicate a vulnerable plaque has been tested. Owing to the pivotal role of lipids and inflammation in the setting of atherosclerosis, most of the biomarkers originated from one of those processes. Conceptually, it is hypothesized that elevated levels of certain lipids will indicate increased susceptibility to the development of unstable atherosclerotic plaque owing to their role in the origin of plaque destabilization. On the other hand, certain molecules can migrate from vulnerable lesion back to circulation, thus creating an opportunity to serve as a biomarker (Figure 1). In addition, recent development of molecular science brought some novel technologies such as microRNA (miRNA).
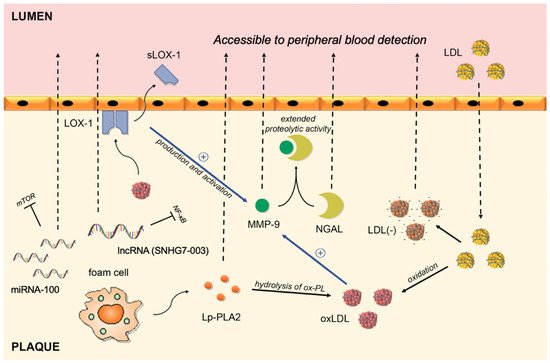
Figure 1. Circulating biomarkers of vulnerable plaques and their interplay in the intima of the coronary artery. LDL migrates from blood to plaque, where it goes through series of modifications. By binding to LOX-1, oxLDL activates and upregulates the production of MMP-9, whereas shedding of the LOX-1 receptor allows its peripheral blood detection. The complex between NGAL and MMP-9 results in extended proteolytic activity of MMP-9. Lp-PLA2 is responsible for hydrolysis of ox-PL on LDL particles and subsequent release of proinflammatory lipids. MMP-9: Matrix metalloproteinase-9; NGAL: Neutrophil gelatinase-associated lipocalin; sLOX-1: soluble part of the Lectin-like oxidized low-density lipoprotein receptor-1; Ox-LDL: oxidized low-density lipoprotein; Ox-PL: oxidized phospholipids; Lp-PLA2: lipoprotein-associated phospholipase A2; LDL(-): electronegative low-density lipoprotein; LDL: low-density lipoprotein; miRNA-100: microRNA-100; lncRNA: long non-coding RNA.
2.1. Inflammation-Based Biomarkers
One of the most extensively studied biomarkers in this setting is high sensitivity C-reactive protein (hs-CRP), an established inflammatory biomarker used in everyday practice worldwide [41]. Aside from the well-known role as the most important acute-phase protein synthetized in the liver, increased CRP levels have been shown to be an independent risk factor for myocardial infarction, predicting CV events better than low-density lipoprotein (LDL) cholesterol [42][43][44]. Studies suggest that apart from the liver, CRP can be synthesized in plaques by macrophages or smooth muscle-like cells [45]. In line with this, Inoue et al. demonstrated CRP is released both from vulnerable coronary plaques and plaques damaged during percutaneous coronary intervention (PCI), whereas Norja et al. demonstrated that CRP immunoreactivity is associated with the progression of atherosclerotic plaque, especially with the vulnerable coronary plaques [46][47]. Apart from representing a biomarker of inflammation, studies suggest that CRP is an effector molecule able to induce a pro-atherothrombotic phenotype in endothelial cells and SMCs and is thus directly implicated in plaque pathophysiology [48]. Although CRP plasma levels undoubtedly reflect the vulnerability of the plaque, perhaps the biggest setback in its clinical implementation in this setting is its low specificity [49][50]. Virtually any inflammatory process in the human body can result in elevated CRP levels. However, CRP could be beneficial as a part of multiple biomarker prognostic score or if used in conjunction with imaging techniques.
Matrix metalloproteinase-9 (MMP-9) belongs to the family of zinc-binding proteolytic enzymes that are capable of degrading most of the extracellular matrix and that take part in all inflammatory processes in humans [51]. Expression of MMP-9 is markedly upregulated in human macrophages stimulated by oxidized low-density lipoprotein (ox-LDL), suggesting its contribution to matrix degradation in the atherosclerotic plaque and making it susceptible to rupture and/or vascular remodeling [52][53]. In line with this, studies on carotid plaques indicate that MMP-9 expression in carotid plaques is higher in vulnerable/symptomatic plaques in comparison to stable ones, whereas serum MMP-9 levels are significantly higher in patients with atheromatous plaques in contrast to patients with fibrous plaques [54][55][56][57]. Furthermore, multiple studies suggest that serum MMP-9 levels are elevated in patients with unstable plaques and Ferroni et al. suggest that MMP-9 serum levels might even provide an index of plaque activity in the setting of coronary artery disease (CAD) [58][59][60]. Notably, certain polymorphisms of MMP-9 confer a susceptibility risk for CAD. The most consistent evidence with respect to association of MMP-9 polymorphisms and CAD is that regarding the C1562T polymorphism. In a recent meta-analysis, Hassanzadeh-Makoui et al. demonstrated that MMP-9 (C1562T) polymorphism was associated with increased risk of CAD susceptibility in the overall analysis, but markedly in the Asian population [61]. The pathophysiological background of this association is the fact that this variant of MMP-9 decreases binding potential of the proteins involved in the inhibition of transcription to the DNA sequence, thus playing a role in orchestrating the transcription activity of MMP-9 [62]. Rather interestingly, this polymorphism exerts a protective role in a wide spectrum of diseases, such as diabetic nephropathy and anterior open bite [63][64]. In a large prospective trial (n = 1127) that included patients with CAD, the association between MMP-9 levels and the risk of fatal CV events showed a hazard ratio of 1.3, even after adjustment for confounders in terms of therapy and other clinical confounders whereas multivariate regression analysis by Ezhov et al. disclosed that MMP-9 is a strong independent predictor of plaque instability in stable CAD patients [65]. An important line of evidence with regard to MMP-9 implementation was brought by Wang et al. who showed that, in patients presenting with unstable angina, serum MMP-9 levels may discriminate patients who have unstable plaques from patients who do not have plaques, directly implicating a viable clinical usefulness of the MMP-9 [66]. However, further well-designed studies are needed to establish a putative role of this marker in everyday clinical setting.
A member of the lipocalin superfamily, Neutrophil gelatinase-associated lipocalin (NGAL) is an important regulator of the MMP-9 enzymatic activity and is thus implicated in progression of atherosclerosis [67]. NGAL creates a complex with MMP-9 which then inhibits degradation of MMP-9, consequently extending its proteolytic activity [67]. Circulating levels of both NGAL and MMP-9/NGAL complexes are significantly increased in asymptomatic patients with vulnerable carotid plaques, as demonstrated by Eilenberg et al. [68]. Multiple authors reported higher NGAL levels in patients with ACS in comparison to patients with stable CAD [69][70]. In addition, NGAL has been shown in several studies to correlate with poorer prognosis and to predict all-cause mortality and major adverse cardiac events (MACE) in patients with ACS [71][72][73]. However, implication of NGAL in a myriad of processes such as acute kidney injury, heart failure, and stable CAD, as well as inconsistence in data regarding prediction of clinical outcomes reduces its chance for establishment as a biomarker of plaque vulnerability [70][74][75].
Another molecule that requires attention is soluble part of the Lectin-like oxidized low-density lipoprotein receptor-1 (sLOX-1), produced by shedding of the LOX-1 [76]. Interaction between ox-LDL and its principal receptor, the LOX-1, appears to play a role in vascular dysfunction, including cells apoptosis and MMP production and activation, evoking the plaque rupture or erosion [77][78][79][80]. LOX-1 is abundantly present advanced in human atherosclerotic lesions. However, the main advantage of LOX-1 in terms of biomarker value is that its soluble part, the sLOX-1, is significantly elevated during the acute stage of ACS whilst not in general acute inflammatory diseases or stable CAD [81][82]. In fact, elevated sLOX-1 levels were detectable at an earlier stage after the onset of ACS in comparison to those of troponin-T (TnT), indicating that sLOX-1 reflects the atherosclerotic plaque vulnerability/rupture even before ischemic cardiac damage becomes clinically evident [81]. Furthermore, sLOX, but neither hsCRP nor hsTnT, can differentiate ACS with plaque rupture from those without, and ACS with thin-cap fibroatheroma from those without, as shown by OCT studies [83]. Additionally, the accuracy of the ACS diagnosis improved when sLOX-1 and hs-TnT were measured in combination [84].
2.2. Lipid-Based Biomarkers
It has so far been well established by large epidemiological studies that higher lipid levels correlate with the occurrence of major CV events [85][86][87]. Hence, since major CV events arise from vulnerable plaques, it is reasonable to infer that lipid status could indicate plaque vulnerability. Unlike in plasma, where LDL can be scarcely modified, in the atherosclerotic plaque, LDL is easily modified under the effect of free radicals and enzymatic activity [88]. More importantly, these modified LDLs gain inflammatory properties and become aggregated, impeding their return to the circulation [84][89]. However, a small portion of these reach plasma and it is therefore hypothesized that these could serve as biomarkers.
Ox-LDL is one of the key pathophysiologic contributors to the atherosclerotic plaque development and progression [90]. It has been demonstrated by multiple authors that amount of plaque ox-LDL correlates with plaque instability, especially in symptomatic carotid artery disease [91][92][93]. However, the translation of experimental evidence in humans with aimed at the demonstration of the association between ox-LDL plasma levels with CV events proved to be difficult as it resulted in contrasting findings [94][95][96][97]. This example highlights the challenging nature of plaque biomarker implementation in clinical practice, as although ox-LDL is a major participant in the in proinflammatory process associated with plaque rupture, its plasma levels do not seem to reliably reflect the vulnerability of the plaque.
Electronegative LDL (LDL(−)) is a modified fraction of the LDL that holds physical and chemical characteristics that differ from those of native LDL, such as increased lipoprotein-associated phospholipase A2 activity (Lp-PLA2), ceramide, clusterine, non-esterified fatty acid content, as well as increased aggregation level [98]. LDL(−) normally comprises around 3−5% of the total LDL plasma quantity, yet in certain CV pathologies this fraction rises [99]. A moderate rise of LDL(−) is commonly observed in patients with classical CV risk factors such as hypercholesterolemia, active smoking, diabetes, and metabolic syndrome [100][101][102][103]. Moreover, LDL(−) is an important mediator of atherogenesis which triggers the detrimental cascade by binding to LOX-1, the aforementioned inflammatory biomarker [104]. It has been recently shown that the most electronegative and the most atherogenic LDL fraction, the L5 fraction, is elevated in plasma of patients in STEMI [105]. In addition, L5 from STEMI patients can enhance platelet aggregation in vitro [106]. L5 levels have been also elevated in patients with ischemic stroke, and Shen et al. argue that L5 could thus serve as a marker of plaque vulnerability in those patients [106]. Even though this biomarker could be promising, a much larger body of well-design studies is needed to confirm it.
Lp-PLA2, originally named platelet-activating factor acetylhydrolase (PAF-AH), is an enzyme formed by macrophages and foam cells in atherosclerotic plaque where it is responsible for hydrolysis of oxidized phospholipids (ox-PL) on LDL particles and subsequent release of proinflammatory lipids [107]. It has been shown that stable atherosclerotic plaques contain only small amount of Lp-PLA2, unlike vulnerable plaques which are abundant in this molecule, markedly inside necrotic core [108]. Importantly, unlike CRP, which is elevated in most inflammatory processes, Lp-PLA2 represents a vascular-specific inflammatory marker and its plasma concentration is stable in terms of time, which largely increases the putative role of this molecule as a biomarker [109]. In addition, Lp-PLA2 level is independent of insulin resistance, unlike most of the other biomarkers [110]. In a study by Sarlon-Bartoli et al., authors demonstrated that patients with confirmed unstable carotid plaque, even if asymptomatic, have higher Lp-PLA2 plasma levels [111]. Similarly, Dong-Ling et al. demonstrated the same in patients with coronary artery plaques, highlighting that the specificity of serum Lp-PLA2 was stronger than that of hs-CRP in this setting [112]. Furthermore, multiple epidemiological studies and meta-analysis demonstrated that Lp-PLA2 is independently associated with the risk of CAD, but rather interestingly, not with atherosclerosis in abdominal aorta [113][114][115]. This discrepancy was further investigated by Fenning et al., which confirmed the differential role of Lp-PLA2 in the inflammatory cascade between different plaques [116]. The authors hypothesize that the observed discrepancy arises from differences in flow hemodynamics and embryologic origin of the vasculature. This discrepancy is very important from our point of view as it highlights the difficulty of using non-cardiac vasculature as a surrogate marker for detection and management of atherosclerotic plaques in coronary arteries. In summary, LpPLA2 seems to be a potentially beneficial adjunctive biomarker as it is both specific for vascular inflammation and, relatively speaking, for CAD.
2.3. Non-Coding RNAs
2.3.1. MiRNAs
MiRNAs are short, single-stranded, non-coding RNA sequences that remain stable and are detectable in peripheral blood owing to inclusion in lipid or lipoprotein complexes [117]. The main function of miRNAs is post-transcriptional regulation of myriad of process in the human body [118]. The first reports on alterations in the expression of various miRNAs in human atherosclerotic plaques were brought by Cipollone et al. [119]. Among numerous muscle-enriched, vascular-enriched, myeloid cell-enriched miRNAs that were tested as markers of coronary plaque vulnerability by Soeki et al., only plasma miRNA-100 levels seem to indicate plaque vulnerability [120]. Authors have drawn this conclusion based on the findings that miRNA-100 levels were higher in coronary sinus blood samples in comparison to aortic blood samples and that transcoronary concentration gradients of circulating miRNA-100 positively correlated with percentage of plaque lipid volume and negatively with percentage of plaque fibrous volume. Soeki et al. argue that miRNA-100 is released into the coronary circulation from lipid-rich plaques as a form of compensatory reaction for plaque stabilization via suppressing the mammalian target of rapamycin signaling pathway. Another interesting miRNA in this setting is miRNA-21, miRNA whose role in atherosclerotic plaque progression has been well-established [121]. As demonstrated by Jin et al., vulnerable plaques and atherosclerotic lesions in apolipoprotein E (ApoE)-deficient mice exhibit lower miRNA-21 levels [122]. Moreover, mice-deficient in both ApoE and miRNA-21 were more prone to rupture, highlighting the implication of miRNA-21 in plaque instability. MiRNA-21 seems to enhance plaque stability by increasing SMC proliferation and consequent stabilization of the SMC-rich fibrous cap that shields the core of the plaque [122]. In a study by He et al., plasma levels of miRNA-21 correlated with several indicators of plaque instability, such as the size of the largest core area, thickness of the fibrous cap, and the lipid pool and the number of macrophages [123]. Several other studies demonstrated that miRNA-21 was upregulated in both patients with ACS and patients with ischemic stroke, as well as in stable CAD [124][125][126]. Apart from miRNA-100, more miRNAs have been recently associated with plaque burden (miR-126, miR-145, miR-155, and miR-29b). The miR-126 is particularly relevant, as its levels are associated with platelet function and can be modulated by antiplatelet treatment. As demonstrated by Carino et al., switching from dual antiplatelet treatment (acetylsalicylic acid +clopidogrel) to ticagrelor is associated with significant modulation in the circulating levels of certain miRNAs, paving the way for the use of circulating miRNAs as biomarkers of platelets activity in response to antiplatelet treatment [127].
However, miRNA technology currently bears some limits in terms of implementation in clinical practice [128]. The problem of normalizing plasma miRNAs levels is important, yet challenging. As Izawa et al. highlight, the expression profile of circulating miRNAs may change according to the current state of the patient, hence “housekeeping” miRNAs has not yet been established [129]. Consequently, a larger body of studies including large cohort of patients are needed to clarify whether circulating miRNAs could be useful as biomarker in this, but in other clinical settings as well.
2.3.2. Long Non-Coding RNA (lncRNA)
LncRNAs were first identified as non-protein-coding RNAs with longer than 200 nucleotides in 1992 [130]. Since then, it has been discovered that by regulating gene expression and functions of other molecules through multiple approaches (epigenetic regulation, transcriptional regulation, post-transcriptional regulation, and many others), these non-coding RNAs have been implicated in a myriad of different pathophysiological processes in the human body [131]. We have been recently acquainted with the key role of lncRNA in inflammatory responses, thus implicating a potential link between lncRNA expression and atherosclerosis [132]. In fact, several studies have established this connection, paving a way for use of lncRNA in both diagnostic and treatment of CAD [133][134][135]. Plaque Enriched Long Noncoding RNA in Atherosclerotic Macrophage Regulation (PELATON) is a recently recognized lncRNA that has been upregulated in unstable atherosclerotic plaques [133]. Hung et al. demonstrated that key atherosclerotic processes of phagocytosis, ox-LDL uptake, and ROS production were all markedly affected by knockdown of PELATON, thus implicating that this lncRNA represents a potential target inhibition of plaque progression [133]. On the other hand, Pan et al. argue that lncRNA-SNHG7-003 has the potential to be a circulation lncRNA biomarker for evaluating plaque stability in patients with CAD, as this lncRNA was validated to be significantly downregulated in blood samples of patients with unstable plaques [134]. The pathophysiological background for this association lies in the ability of SNHG7-003 to inhibit LPS-induced activation of NF-κB pathway, thus regulating inflammatory response in human monocytes and macrophages [134]. Relevant biomarkers that have the potential to be used in the diagnosis of vulnerable plaques are presented in the Table 1.
Table 1. Biomarkers in diagnostic approach to vulnerable plaque.
| Biomarker | Pathophysiological Pathway | Supporting Evidence |
|---|---|---|
| hs-CRP | An acute phase protein that, apart from liver, can be synthesized in plaques by macrophages or smooth muscle-like cells | Inoue et al. [46] Norja et al. [47] |
| MMP-9 | A proteolytic enzyme capable of degrading the extracellular matrix; upregulated in human macrophages stimulated by ox-LDL | Ezhov et al. [65] Wang et al. [66] |
| NGAL | Creates a complex with MMP-9 that inhibits degradation of MMP-9, thus extending its proteolytic activity | Eilenberg et al. [68] |
| sLOX-1 | A soluble form of LOX-1 receptor; interaction between ox-LDL (ligand) and LOX-1 (receptor) plays a role in vascular dysfunction | Hayashida et al. [81] Ueda et al. [82] Kobayashi et al. [83] |
| Ox-LDL | An oxidized fraction of the LDL; major participant in the proinflammatory processes associated with plaque rupture | Wang et al. [91] Wang et al. [92] Sigala et al. [93] |
| Electronegative LDL | A modified fraction of the LDL; physical and chemical characteristics differ from native LDL (increased Lp-PLA2 activity, ceramide, clusterine, non-esterified fatty acid content, increased aggregation level) | Lu et al. [104] Yang et al. [105] |
| Lp-PLA2 | An enzyme formed by macrophages and foam cells; hydrolyses oxidized phospholipids on LDL particles and subsequently releases proinflammatory lipids | Sarlon-Bartoli et al. [111] Dong-Ling et al. [112] Fenning et al. [116] |
| MicroRNA-100 | Post-transcriptional regulation | Soeki et al. [120] |
| MicroRNA-21 | Post-transcriptional regulation | Jin et al. [122] He et al. [123] |
hs-CRP: high sensitivity C-reactive protein; MMP-9: Matrix metalloproteinase-9; NGAL: Neutrophil gelatinase-associated lipocalin; sLOX-1: soluble part of the Lectin-like oxidized low-density lipoprotein receptor-1; Electronegative LDL: electronegative low-density lipoprotein; Ox-LDL: oxidized low-density lipoprotein; Lp-PLA2: lipoprotein-associated phospholipase A2.
3. Role of Invasive Imaging Methods in Plaque Pathobiology Assessment
Invasive coronary imaging methods, such as IVUS, have limited accuracy in identifying plaques likely to become culprit lesions [136][137][138]. In addition, as discussed by Bourantas et al., there are several other limitations that burden the use of those methods in vulnerable plaque detection [138]. As we previously mentioned, currently the most important invasive imaging modality is the intravascular OCT. OCT seems to be a powerful tool for discrimination of three types of unstable plaque morphologies underlying coronary thrombosis—such as plaque rupture, erosion, and calcified nodules—but also plaque healing [139]. All the pros and cons of OCT versus IVUS were discussed in a review by Su et al. [140]. The main advantage of OCT in comparison to IVUS is 10× higher resolution (15–20 µm vs. 150–200 µm) [141]. The discrepancy in resolution is also clinically relevant, as Lv et al. demonstrated that there are significant differences between IVUS and OCT plaque cap thickness measurements, the latter reflecting the plaque morphology more accurate [141]. Accurate plaque thickness is important as it can be used to predict plaque growth and vulnerability. On the other hand, OCT is characterized by lower tissue penetration than IVUS [141]. In addition, the development of hybrid intravascular imaging methods, which are currently undergoing preclinical evaluation, seem to enable more complete evaluation of the plaque pathophysiology [142]. In fact, there is an ongoing study, the ILUMIEN IV study, that is planned to be completed in 2022, and that will determine if OCT-guided stenting will provide better clinical outcomes than the angiographic guidance alone [140]. Additionally, near-infrared spectroscopy (NIRS) emerged as a viable method in this setting, as multiple studies have demonstrated that the detection of lipids in the walls of the coronary vessels identifies vulnerable patients at increased risk of new CV event [141]. Moreover, recent research regarding the combination of OCT and NIRS in plaque detection yielded promising results [142]. However, further large-scale trials are needed in order to support these notions.
References
- Simionescu, M. Implications of early structural-functional changes in the endothelium for vascular disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 266–274.
- Aguilar-Ballester, M.; Herrero-Cervera, A.; Vinué, Á.; Martínez-Hervás, S.; González-Navarro, H. Impact of Cholesterol Metabolism in Immune Cell Function and Atherosclerosis. Nutrients 2020, 12, 2021.
- Sorci-Thomas, M.G.; Thomas, M.J. Microdomains, Inflammation, and Atherosclerosis. Circ. Res. 2016, 118, 679–691.
- Crea, F.; Libby, P. Acute Coronary Syndromes: The Way Forward from Mechanisms to Precision Treatment. Circulation 2017, 136, 1155–1166.
- Kumrić, M.; Tičinović Kurir, T.; Borovac, J.A.; Božić, J. The Role of Natural Killer (NK) Cells in Acute Coronary Syndrome: A Comprehensive Review. Biomolecules 2020, 10, 1514.
- Tabas, I.; Tall, A.; Accili, D. The impact of macrophage insulin resistance on advanced atherosclerotic plaque progression. Circ. Res. 2010, 106, 58–67.
- Tabas, I.; Williams, K.J.; Boren, J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: Update and therapeutic implications. Circulation 2007, 116, 1832–1844.
- Kirichenko, T.V.; Markina, Y.V.; Sukhorukov, V.N.; Khotina, V.A.; Wu, W.K.; Orekhov, A.N. A Novel Insight at Atherogenesis: The Role of Microbiome. Front. Cell Dev. Biol. 2020, 8, 586189.
- Doran, A.C.; Meller, N.; McNamara, C.A. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 812–819.
- Tuttolomondo, A.; Di Raimondo, D.; Pecoraro, R.; Arnao, V.; Pinto, A.; Licata, G. Atherosclerosis as an inflammatory disease. Curr. Pharm Des. 2012, 18, 4266–4288.
- Virmani, R.; Burke, A.P.; Kolodgie, F.D.; Farb, A. Vulnerable plaque: The pathology of unstable coronary lesions. J. Interv. Cardiol. 2002, 15, 439–446.
- Hafiane, A. Vulnerable Plaque, Characteristics, Detection, and Potential Therapies. J. Cardiovasc. Dev. Dis. 2019, 6, 26.
- Borovac, J.A.; Glavas, D.; Susilovic Grabovac, Z.; Supe Domic, D.; D’Amario, D.; Bozic, J. Catestatin in Acutely Decompensated Heart Failure Patients: Insights from the CATSTAT-HF Study. J. Clin. Med. 2019, 8, 1132.
- Libby, P.; Pasterkamp, G. Requiem for the ‘vulnerable plaque’. Eur. Heart, J. 2015, 36, 2984–2987.
- Stefanadis, C.; Antoniou, C.K.; Tsiachris, D.; Pietri, P. Coronary Atherosclerotic Vulnerable Plaque: Current Perspectives. J. Am. Heart Assoc. 2017, 6, e005543.
- Chang, H.J.; Lin, F.Y.; Lee, S.E.; Andreini, D.; Bax, J.; Cademartiri, F.; Chinnaiyan, K.; Chow, B.J.W.; Conte, E.; Cury, R.C.; et al. Coronary Atherosclerotic Precursors of Acute Coronary Syndromes. J. Am. Coll. Cardiol. 2018, 71, 2511–2522.
- Arbustini, E.; Dal Bello, B.; Morbini, P.; Burke, A.P.; Bocciarelli, M.; Specchia, G.; Virmani, R. Plaque erosion is a major substrate for coronary thrombosis in acute myocardial infarction. Heart 1999, 82, 269–272.
- Kolodgie, F.D.; Burke, A.P.; Farb, A.; Weber, D.K.; Kutys, R.; Wight, T.N.; Virmani, R. Differential accumulation of proteoglycans and hyaluronan in culprit lesions: Insights into plaque erosion. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1642–1648.
- Bilalic, A.; Ticinovic Kurir, T.; Kumric, M.; Borovac, J.A.; Matetic, A.; Supe-Domic, D.; Bozic, J. Circulating Levels of Dephosphorylated-Uncarboxylated Matrix Gla Protein in Patients with Acute Coronary Syndrome. Molecules 2021, 26, 1108.
- Libby, P.; Pasterkamp, G.; Crea, F.; Jang, I.K. Reassessing the Mechanisms of Acute Coronary Syndromes. Circ. Res. 2019, 124, 150–160.
- Scalone, G.; Niccoli, G.; Refaat, H.; Vergallo, R.; Porto, I.; Leone, A.M.; Burzotta, F.; D’Amario, D.; Liuzzo, G.; Fracassi, F.; et al. Not all plaque ruptures are born equal: An optical coherence tomography study. Eur. Heart J. Cardiovasc. Imaging. 2017, 18, 1271–1277.
- Niccoli, G.; Montone, R.A.; Cataneo, L.; Cosentino, N.; Gramegna, M.; Refaat, H.; Porto, I.; Burzotta, F.; Trani, C.; Leone, A.M.; et al. Morphological-biohumoral correlations in acute coronary syndromes: Pathogenetic implications. Int. J. Cardiol. 2014, 171, 463–466.
- Jia, H.; Abtahian, F.; Aguirre, A.D.; Lee, S.; Chia, S.; Lowe, H.; Kato, K.; Yonetsu, T.; Vergallo, R.; Hu, S.; et al. In vivo diagnosis of plaque erosion and calcified nodule in patients with acute coronary syndrome by intravascular optical coherence tomography. J. Am. Coll. Cardiol. 2013, 62, 1748–1758.
- Borovac, J.A.; D’Amario, D.; Vergallo, R.; Porto, I.; Bisignani, A.; Galli, M.; Annibali, G.; Montone, R.A.; Leone, A.M.; Niccoli, G.; et al. Neoatherosclerosis after drug-eluting stent implantation: A novel clinical and therapeutic challenge. Eur. Heart J. Cardiovasc. Pharmacother. 2019, 5, 105–116.
- Henson, P.M.; Bratton, D.L.; Fadok, V.A. Apoptotic cell removal. Curr. Biol. 2001, 11, R795–R805.
- Kojima, Y.; Volkmer, J.P.; McKenna, K.; Civelek, M.; Lusis, A.J.; Miller, C.L.; Direnzo, D.; Nanda, V.; Ye, J.; Connolly, A.J. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature 2016, 536, 86–90.
- Sather, S.; Kenyon, K.D.; Lefkowitz, J.B.; Liang, X.; Varnum, B.C.; Henson, P.M.; Graham, D.K. A soluble form of the Mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood 2007, 109, 1026–1033.
- Bäck, M.; Yurdagul, A., Jr.; Tabas, I.; Öörni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406.
- Serhan, C.N. Novel lipid mediators and resolution mechanisms in acute inflammation: To resolve or not? Am. J. Pathol. 2010, 177, 1576–1591.
- Tabas, I.; Glass, C.K. Anti-inflammatory therapy in chronic disease: Challenges and opportunities. Science 2013, 339, 166–172.
- Tabas, I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 2010, 10, 36–46.
- Fredman, G.; Hellmann, J.; Proto, J.D.; Kuriakose, G.; Colas, R.A.; Dorweiler, B.; Connolly, E.S.; Solomon, R.; Jones, D.M.; Heyer, E.J. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat. Commun. 2016, 7, 12859.
- Viola, J.; Lemnitzer, P.; Jansen, Y.; Csaba, G.; Winter, C.; Neideck, C.; Silvestre-Roig, C.; Dittmar, G.; Döring, Y.; Drechsler, M. Resolving lipid mediators maresin 1 and resolvin D2 prevent atheroprogression in mice. Circ.Res. 2016, 119, 1030–1038.
- Fredman, G.; Kamaly, N.; Spolitu, S.; Milton, J.; Ghorpade, D.; Chiasson, R.; Kuriakose, G.; Perretti, M.; Farokzhad, O.; Tabas, I. Targeted nanoparticles containing the pro-resolving peptide Ac2-26 protect against advanced atheosclerosis in hypercholesterolemic mice. Sci. Transl. Med. 2015, 7, 275ra20.
- Tabas, I. 2016 Russell Ross Memorial Lecture in Vascular Biology: Molecular-Cellular Mechanisms in the Progression of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 183–189.
- Fredman, G.; Ozcan, L.; Spolitu, S.; Hellmann, J.; Spite, M.; Backs, J.; Tabas, I. Resolvin D1 limits 5-lipoxygenase nuclear localization and leukotriene B4 synthesis by inhibiting a calcium-activated kinase pathway. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 14530–14535.
- Dalli, J.; Serhan, C. Specific lipid mediator signatures of human phagocytes: Microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood 2012.
- Cai, B.; Thorp, E.B.; Doran, A.C.; Subramanian, M.; Sansbury, B.E.; Lin, C.S.; Spite, M.; Fredman, G.; Tabas, I. MerTK cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 6526–6531.
- Peeters, W.; Hellings, W.E.; de Kleijn, D.P.; de Vries, J.P.; Moll, F.L.; Vink, A.; Pasterkamp, G. Carotid atherosclerotic plaques stabilize after stroke: Insights into the natural process of atherosclerotic plaque stabilization. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 128–133.
- Ylä-Herttuala, S.; Bentzon, J.F.; Daemen, M.; Falk, E.; Garcia-Garcia, H.M.; Herrmann, J.; Hoefer, I.; Jauhiainen, S.; Jukema, J.W.; Krams, R. ESC Working Group of Atherosclerosis and Vascular Biology. Stabilization of atherosclerotic plaques: An update. Eur. Heart J. 2013, 34, 3251–3258.
- Moutachakkir, M.; Lamrani Hanchi, A.; Baraou, A.; Boukhira, A.; Chellak, S. Immunoanalytical characteristics of C-reactive protein and high sensitivity C-reactive protein. Ann Biol. Clin. (Paris). 2017, 75, 225–229, English.
- Li, Y.; Zhong, X.; Cheng, G.; Zhao, C.; Zhang, L.; Hong, Y.; Wan, Q.; He, R.; Wang, Z. Hs-CRP and all-cause, cardiovascular, and cancer mortality risk: A meta-analysis. Atherosclerosis 2017, 259, 75–82.
- Ferreiros, E.R.; Boissonnet, C.P.; Pizarro, R.; Merletti, P.F.; Corrado, G.; Cagide, A.; Bazzino, O.O. Independent prognostic value of elevated C-reactive protein in unstable angina. Circulation 1999, 100, 1958–1963.
- Lagrand, W.K.; Visser, C.A.; Hermens, W.T.; Niessen, H.W.; Verheugt, F.W.; Wolbink, G.J.; Hack, C.E. C-reactive protein as a cardiovascular risk factor: More than an epiphenomenon? Circulation 1999, 100, 96–102.
- Ishikawa, T.; Hatakeyama, K.; Imamura, T.; Date, H.; Shibata, Y.; Hikichi, Y.; Asada, Y.; Eto, T. Involvement of C-reactive protein obtained by directional coronary atherectomy in plaque instability and developing restenosis in patients with stable or unstable angina pectoris. Am. J. Cardiol. 2003, 91, 287–292.
- Inoue, T.; Kato, T.; Uchida, T.; Sakuma, M.; Nakajima, A.; Shibazaki, M.; Imoto, Y.; Saito, M.; Hashimoto, S.; Hikichi, Y.; et al. Local release of C-reactive protein from vulnerable plaque or coronary arterial wall injured by stenting. J. Am. Coll. Cardiol. 2005, 46, 239–245.
- Norja, S.; Nuutila, L.; Karhunen, P.J.; Goebeler, S. C-reactive protein in vulnerable coronary plaques. J. Clin. Pathol. 2007, 60, 545–548.
- Cirillo, P.; Golino, P.; Calabrò, P.; Calì, G.; Ragni, M.; De Rosa, S.; Cimmino, G.; Pacileo, M.; De Palma, R.; Forte, L.; et al. C-reactive protein induces tissue factor expression and promotes smooth muscle and endothelial cell proliferation. Cardiovasc. Res. 2005, 68, 47–55.
- S Hackam, D.G.; Shumak, S.L. C-reactive protein for the prediction of cardiovascular risk: Ready for prime-time? CMAJ. 2004, 170, 1563–1565.
- Sabatine, M.S.; Morrow, D.A.; Jablonski, K.A.; Rice, M.M.; Warnica, J.W.; Domanski, M.J.; Hsia, J.; Gersh, B.J.; Rifai, N.; Ridker, P.M.; et al. Prognostic significance of the Centers for Disease Control/American Heart Association high-sensitivity C-reactive protein cut points for cardiovascular and other outcomes in patients with stable coronary artery disease. Circulation 2007, 115, 1528–1536.
- Johnson, J.L. Matrix metalloproteinases: Influence on smooth muscle cells and atherosclerotic plaque stability. Expert Rev. Cardiovasc. Ther. 2007, 5, 265–282.
- Xu, X.P.; Meisel, S.R.; Ong, J.M.; Kaul, S.; Cercek, B.; Rajavashisth, T.B.; Sharifi, B.; Shah, P.K. Oxidized low-density lipoprotein regulates matrix metallo- proteinase-9 and its tissue inhibitor in human monocyte-derived macrophages. Circulation 1999, 99, 993–998.
- Kalela, A.; Koivu, T.A.; Sisto, T.; Kanervisto, J.; Hoyhtya, M.; Sillanaukee, P.; Lehtimaki, T.; Nikkari, S.T. Serum matrix met-alloproteinase-9 concentration in angiographically assessed coronary artery disease. Scand J. Clin Lab Invest 2002, 62, 337–342.
- Guo, Z.Y.; Zhang, B.; Yan, Y.H.; Gao, S.S.; Liu, J.J.; Xu, L.; Hui, P.J. Specific matrix metalloproteinases and calcification factors are associated with the vulnerability of human carotid plaque. Exp. Ther. Med. 2018, 16, 2071–2079.
- Langley, S.R.; Willeit, K.; Didangelos, A.; Matic, L.P.; Skroblin, P.; Barallobre-Barreiro, J.; Lengquist, M.; Rungger, G.; Kapustin, A.; Kedenko, L.; et al. Extracellular matrix proteomics identifies molecular signature of symptomatic carotid plaques. J. Clin. Investig. 2017, 127, 1546–1560.
- Sukhova, G.K.; Schonbeck, U.; Rabkin, E.; Schoen, F.J.; Poole, R.; Billinghurst, R.C.; Libby, P. Evidence for increased collagenolysis by interstitial collagenases-1 and -3 in vulnerable human atheromatous plaques. Circulation 1999, 99, 2503–2509.
- Sluijter, J.P.G.; Pulskens, W.P.C.; Schoneveld, A.H.; Velema, E.; Strijder, C.F.; Moll, F.; de Vries, J.P.; Verheijen, J.; Hanemaaijer, R.; de Kleijn, D.P.; et al. Matrix metalloproteinase 2 is associated with stable and matrix metalloproteinases 8 and 9 with vulnerable carotid atherosclerotic lesions-a study in hu-manendarterectomy specimen pointing to a role for different extracellular matrix metalloproteinase inducer glycosylation forms. Stroke 2006, 37, 235–239.
- Kai, H.; Ikeda, H.; Yasukawa, H.; Kai, M.; Seki, Y.; Kuwahara, F.; Ueno, T.; Sugi, K.; Imaizumi, T. Peripheral blood levels of matrix metalloproteases-2 and -9 are elevated in patients with acute coronary syndromes. J. Am. Coll. Cardiol. 1998, 32, 368–372.
- Fukuda, D.; Shimada, K.; Tanaka, A.; Kusuyama, T.; Yamashita, H.; Ehara, S.; Nakamura, Y.; Kawarabayashi, T.; Iida, H.; Yoshiyama, M.; et al. Comparison of Levels of Serum Matrix Metalloproteinase-9 in Patients With Acute Myocardial Infarction Versus Unstable Angina Pectoris Versus Stable Angina Pectoris. Am. J. Cardiol. 2006, 97, 175–180.
- Ferroni, P.; Basili, S.; Martini, F.; Cardarello, C.M.; Ceci, F.; Di Franco, M.; Bertazzoni, G.; Gazzaniga, P.P.; Alessandri, C. Serum Metalloproteinase 9 Levels in Patients with Coronary Artery Disease: A Novel Marker of Inflammation. J. Investig. Med. 2003, 51, 295–300.
- Hassanzadeh-Makoui, R.; Razi, B.; Aslani, S.; Imani, D.; Tabaee, S.S. The association between Matrix Metallo-proteinases-9 (MMP-9) gene family polymorphisms and risk of Coronary Artery Disease (CAD): A systematic review and meta-analysis. BMC Cardiovasc. Disord. 2020, 20, 1–15.
- Cho, H.-J.; Chae, I.-H.; Park, K.-W.; Ju, J.-R.; Oh, S.; Lee, M.-M.; Park, Y.-B. Functional polymorphism in the promoter region of the gelatinase B gene in relation to coronary artery disease and restenosis after percutaneous coronary intervention. J. Hum. Genet. 2002, 47, 88–91.
- Xie, Y.; Wang, Z.; Chang, L.; Chen, G. Association of MMP-9 polymorphisms with diabetic nephropathy risk. Medicine 2020, 99, e22278.
- Küchler, E.C.; Barreiros, D.; Da Silva, R.O.; De Abreu, J.G.B.; Teixeira, E.C.; Da Silva, R.A.B.; Da Silva, L.A.B.; Filho, P.N.; Romano, F.L.; Granjeiro, J.M.; et al. Genetic Polymorphism in MMP9 May Be Associated With Anterior Open Bite in Children. Braz. Dent. J. 2017, 28, 277–280.
- Blankenberg, S.; Rupprecht, H.J.; Poirier, O.; Bickel, C.; Smieja, M.; Hafner, G.; Meyer, J.; Cambien, F.; Tiret, L. Plasma Concentrations and Genetic Variation of Matrix Metalloproteinase 9 and Prognosis of Patients With Cardiovascular Disease. Circulation 2003, 107, 1579–1585.
- Wang, L.-X.; Lü, S.-Z.; Zhang, W.-J.; Song, X.-T.; Chen, H.; Zhang, L.-J. Comparision of high sensitivity C-reactive protein and matrix metalloproteinase 9 in patients with unstable angina between with and without significant coronary artery plaques. Chin. Med. J. 2011, 124, 1657–1661.
- Sivalingam, Z.; Larsen, S.B.; Grove, E.L.; Hvas, A.-M.; Kristensen, S.D.; Magnusson, N.E. Neutrophil gelatinase-associated lipocalin as a risk marker in cardiovascular disease. Clin. Chem. Lab. Med. 2017, 56, 5–18.
- Eilenberg, W.; Stojkovic, S.; Kaider, A.; Piechota-Polanczyk, A.; Nanobachvili, J.; Domenig, C.M.; Wojta, J.; Huk, I.; Demyanets, S.; Neumayer, C. Neutrophil Gelatinase Associated Lipocalin (NGAL) for Identification of Unstable Plaques in Patients with Asymptomatic Carotid Stenosis. Eur. J. Vasc. Endovasc. Surg. 2019, 57, 768–777.
- Sahinarslan, A.; Kocaman, S.A.; Bas, D.; Akyel, A.; Ercin, U.; Zengin, O.; Timurkaynak, T. Plasma neutrophil gelatinase-associated lipocalin levels in acute myocardial infarction and stable coronary artery disease. Coron. Artery Dis. 2011, 22, 333–338.
- Choi, K.M.; Lee, J.S.; Kim, E.J.; Baik, S.H.; Seo, H.S.; Choi, D.S.; Oh, D.J.; Park, C.G. Implication of lipocalin-2 and visfatin levels in patients with coronary heart disease. Eur. J. Endocrinol. 2008, 158, 203–207.
- Giurgea, G.-A.; Zlabinger, K.; Gugerell, A.; Lukovic, D.; Syeda, B.; Mandic, L.; Pavo, N.; Mester-Tonczar, J.; Traxler-Weidenauer, D.; Spannbauer, A.; et al. Multimarker Approach to Identify Patients with Coronary Artery Disease at High Risk for Subsequent Cardiac Adverse Events: The Multi-Biomarker Study. Biomolecules 2020, 10, 909.
- Lindberg, S.; Pedersen, S.H.; Mogelvang, R.; Jensen, J.S.; Flyvbjerg, A.; Galatius, S.; Magnusson, N.E. Prognostic Utility of Neutrophil Gelatinase-Associated Lipocalin in Predicting Mortality and Cardiovascular Events in Patients With ST-Segment Elevation Myocardial Infarction Treated With Primary Percutaneous Coronary Intervention. J. Am. Coll. Cardiol. 2012, 60, 339–345.
- Zykov, M.Z.; Kashtalap, V.K.; Bykova, I.B.; Hryachkova, O.H.; Kalaeva, V.K.; Shafranskaya, K.S.; Karetnikova, V.K.; Barbarash, O.B. Institute for Complex Problems of Сardiovascular Disease Clinical and Prognostic Value of Serum Neutrophil Gelatinase-Associated Lipocalin in Patients With ST-Segment Elevation Myocardial Infarction. Kardiologiia 2016, 56, 24–29.
- Woitas, R.P.; Scharnagl, H.; Kleber, M.E.; Delgado, G.E.; Grammer, T.B.; Pichler, M.; Krämer, B.K.; März, W.; Stojakovic, T. Neutrophil gelatinase-associated lipocalin levels are U-shaped in the Ludwigshafen Risk and Cardiovascular Health (LURIC) study—Impact for mortality. PLoS ONE 2017, 12, e0171574.
- Kumrić, M.; Borovac, J.; Kurir, T.; Božić, J. Clinical Implications of Uric Acid in Heart Failure: A Comprehensive Review. Life 2021, 11, 53.
- Murase, T.; Kume, N.; Kataoka, H.; Minami, M.; Sawamura, T.; Masaki, T.; Kita, T. Identification of Soluble Forms of Lectin-Like Oxidized LDL Receptor-1. Arter. Thromb. Vasc. Biol. 2000, 20, 715–720.
- Kume, N.; Kita, T. New scavenger receptors and their functions in atherogenesis. Curr. Atheroscler. Rep. 2002, 4, 253–257.
- Kataoka, H.; Kume, N.; Miyamoto, S.; Minami, M.; Morimoto, M.; Hayashida, K.; Hashimoto, N.; Kita, T. Oxidized LDL Modulates Bax/Bcl-2 Through the Lectinlike Ox-LDL Receptor-1 in Vascular Smooth Muscle Cells. Arter. Thromb. Vasc. Biol. 2001, 21, 955–960.
- Kume, N.; Kita, T. Apoptosis of vascular cells by oxidized LDL: Involvement of caspases and LOX-1, and its implication in atherosclerotic plaque rupture. Circ. Res. 2004, 94, 269–270.
- Li, D.; Liu, L.; Chen, H.; Sawamura, T.; Ranganathan, S.; Mehta, J.L. LOX-1 Mediates Oxidized Low-Density Lipoprotein-Induced Expression of Matrix Metalloproteinases in Human Coronary Artery Endothelial Cells. Circulation 2003, 107, 612–617.
- Hayashida, K.; Kume, N.; Murase, T.; Minami, M.; Nakagawa, D.; Inada, T.; Minami, M.; Nakagawa, D.; Inada, T.; Tanaka, M.; et al. Serum soluble lectin-like oxidized low-density lipoprotein receptor-1 levels are elevated in acute coronary syndrome: A novel marker for early diagnosis. Circulation 2005, 112, 812–818.
- Ueda, A.; Kume, N.; Hayashida, K.; Inui-Hayashida, A.; Asai, M.; Kita, T.; Kominami, G. ELISA for Soluble Form of Lectin-Like Oxidized LDL Receptor-1, A Novel Marker of Acute Coronary Syndrome. Clin. Chem. 2006, 52, 1210–1211.
- Kobayashi, N.; Takano, M.; Hata, N.; Kume, N.; Yamamoto, M.; Yokoyama, S.; Shinada, T.; Tomita, K.; Shirakabe, A.; Otsuka, T.; et al. Soluble lectin-like oxidized LDL receptor-1 (sLOX-1) as a valuable diagnostic marker for rupture of thin-cap fibroatheroma: Verification by optical coherence tomography. Int. J. Cardiol. 2013, 168, 3217–3223.
- Kobayashi, N.; Hata, N.; Kume, N.; Shinada, T.; Tomita, K.; Shirakabe, A.; Kitamura, M.; Nozaki, A.; Inami, T.; Seino, Y.; et al. Soluble Lectin-Like Oxidized LDL Receptor-1 and High-Sensitivity Troponin T as Diagnostic Biomarkers for Acute Coronary Syndrome–Improved Values With Combination Usage in Emergency Rooms–. Circ. J. 2011, 75, 2862–2871.
- Gu, X.; Li, Y.; Chen, S.; Yang, X.; Liu, F.; Li, Y.; Li, J.; Cao, J.; Liu, X.; Chen, J.; et al. Association of Lipids With Ischemic and Hemorrhagic Stroke. Stroke 2019, 50, 3376–3384.
- Ma, Z.; Yue, Y.; Luo, Y.; Wang, W.; Cao, Y.; Fang, Q. Clinical Utility of the Inflammatory Factors Combined With Lipid Markers in the Diagnostic and Prognostic Assessment of Ischemic Stroke: Based on Logistic Regression Models. J. Stroke Cerebrovasc. Dis. 2020, 29, 104653.
- Claessen, B.E.; Guedeney, P.; Gibson, C.M.; Angiolillo, D.J.; Cao, D.; Lepor, N.; Mehran, R. Lipid Management in Patients Presenting With Acute Coronary Syndromes: A Review. J. Am. Hear. Assoc. 2020, 9, e018897.
- Ke, L.-Y.; Law, S.H.; Mishra, V.K.; Parveen, F.; Chan, H.-C.; Lu, Y.-H.; Chu, C.-S. Molecular and Cellular Mechanisms of Electronegative Lipoproteins in Cardiovascular Diseases. Biomedicines 2020, 8, 550.
- Lehti, S.; Nguyen, S.D.; Belevich, I.; Vihinen, H.; Heikkilä, H.M.; Soliymani, R.; Käkelä, R.; Saksi, J.; Jauhiainen, M.; Grabowski, G.A.; et al. Extracellular Lipids Accumulate in Human Carotid Arteries as Distinct Three-Dimensional Structures and Have Proinflammatory Properties. Am. J. Pathol. 2018, 188, 525–538.
- Hartley, A.; Haskard, D.; Khamis, R. Oxidized LDL and anti-oxidized LDL antibodies in atherosclerosis – Novel insights and future directions in diagnosis and therapy. Trends Cardiovasc. Med. 2019, 29, 22–26.
- Wang, A.; Yang, Y.; Su, Z.; Yue, W.; Hao, H.; Ren, L.; Wang, Y.; Cao, Y.; Wang, Y. Association of Oxidized Low-Density Lipoprotein With Prognosis of Stroke and Stroke Subtypes. Stroke 2017, 48, 91–97.
- Wang, A.; Dai, L.; Zhang, N.; Lin, J.; Chen, G.; Zuo, Y.; Li, H.; Wang, Y.; Meng, X.; Wang, Y. Oxidized low-density lipoprotein (LDL) and LDL cholesterol are associated with outcomes of minor stroke and TIA. Atherosclerosis 2020, 297, 74–80.
- Sigala, F.; Kotsinas, A.; Savari, P.; Filis, K.; Markantonis, S.; Iliodromitis, E.K.; Gorgoulis, V.G.; Andreadou, I. Oxidized LDL in human carotid plaques is related to symptomatic carotid disease and lesion instability. J. Vasc. Surg. 2010, 52, 704–713.
- Yan, Z.; Fu, B.; He, D.; Zhang, Y.; Liu, J.; Zhang, X. The relationship between oxidized low-density lipoprotein and related ratio and acute cerebral infarction. Medicine 2018, 97, e12642.
- Zhang, Y.-C.; Tang, Y.; Chen, Y.; Huang, X.-H.; Zhang, M.; Chen, J.; Sun, Y.-G.; Li, Y.-G. Oxidized Low-Density Lipoprotein and C-Reactive Protein Have Combined Utility for Better Predicting Prognosis After Acute Coronary Syndrome. Cell Biophys. 2013, 68, 379–385.
- Wilson, P.W.; Ben-Yehuda, O.; McNamara, J.; Massaro, J.; Witztum, J.; Reaven, P.D. Autoantibodies to oxidized LDL and cardiovascular risk: The Framingham Offspring Study. Atherosclerosis 2006, 189, 364–368.
- Holvoet, P.; Kritchevsky, S.B.; Tracy, R.P.; Mertens, A.; Rubin, S.M.; Butler, J.; Goodpaster, B.; Harris, T.B. The metabolic syndrome, circulating oxidized LDL, and risk of myocardial infarction in well-functioning elderly people in the health, aging, and body composition cohort. Diabetes 2004, 53, 1068–1073.
- Rivas-Urbina, A.; Rull, A.; Ordóñez-Llanos, J.; Sanchez-Quesada, J.L. Electronegative LDL: An Active Player in Atherogenesis or a By- Product of Atherosclerosis? Curr. Med. Chem. 2019, 26, 1665–1679.
- Chu, C.-S.; Law, S.H.; Lenzen, D.; Tan, Y.-H.; Weng, S.-F.; Ito, E.; Wu, J.-C.; Chen, C.-H.; Chan, H.-C.; Ke, L.-Y. Clinical Significance of Electronegative Low-Density Lipoprotein Cholesterol in Atherothrombosis. Biomedicine 2020, 8, 254.
- Hsu, J.-F.; Chou, T.-C.; Lu, J.; Chen, S.-H.; Chen, F.-Y.; Chen, C.-C.; Chen, J.L.; Elayda, M.; Ballantyne, C.M.; Shayani, S.; et al. Low-Density Lipoprotein Electronegativity Is a Novel Cardiometabolic Risk Factor. PLoS ONE 2014, 9, e107340.
- Chang, C.-T.; Shen, M.-Y.; Lee, A.-S.; Wang, C.-C.; Chen, W.-Y.; Chang, C.-M.; Chang, K.-C.; Stancel, N.; Chen, C.-H. Electronegative low-density lipoprotein increases the risk of ischemic lower-extremity peripheral artery disease in uremia patients on maintenance hemodialysis. Sci. Rep. 2017, 7, 4654.
- Lai, Y.-S.; Yang, T.-C.; Chang, P.-Y.; Chang, S.-F.; Ho, S.-L.; Chen, H.-L.; Lu, S.-C. Electronegative LDL is linked to high-fat, high-cholesterol diet–induced nonalcoholic steatohepatitis in hamsters. J. Nutr. Biochem. 2016, 30, 44–52.
- Estruch, M.; Miñambres, I.; Sanchez-Quesada, J.L.; Soler, M.; Pérez, A.; Ordoñez-Llanos, J.; Benitez, S. Increased inflammatory effect of electronegative LDL and decreased protection by HDL in type 2 diabetic patients. Atherosclerosis 2017, 265, 292–298.
- Lu, J.; Yang, J.H.; Burns, A.R.; Tang, D.; Walterscheid, J.P.; Suzuki, S.; Yang, C.Y.; Sawamura, T.; Chen, C.H. Mediation of electronegative low-density lipoprotein signaling by LOX-1: A possible mechanism of endothelial apoptosis. Circ. Res. 2009, 104, 619–627.
- Yang, T.-C.; Chang, P.-Y.; Lu, S.-C. L5-LDL from ST-elevation myocardial infarction patients induces IL-1β production via LOX-1 and NLRP3 inflammasome activation in macrophages. Am. J. Physiol. Circ. Physiol. 2017, 312, H265–H274.
- Shen, M.-Y.; Chen, F.-Y.; Hsu, J.-F.; Fu, R.-H.; Chang, C.-M.; Chang, C.-T.; Liu, C.-H.; Wu, J.-R.; Lee, A.-S.; Chan, H.-C.; et al. Plasma L5 levels are elevated in ischemic stroke patients and enhance platelet aggregation. Blood 2016, 127, 1336–1345.
- Bonnefont-Rousselot, D. La Lp-PLA2, marqueur d’inflammation vasculaire et de vulnérabilité de la plaque d’athérosclérose. Annales Pharmaceutiques Françaises 2016, 74, 190–197.
- Kolodgie, F.D.; Burke, A.P.; Skorija, K.S.; Ladich, E.; Kutys, R.; Makuria, A.T.; Virmani, R. Lipoprotein-Associated Phospholipase A 2 Protein Expression in the Natural Progression of Human Coronary Atherosclerosis. Arter. Thromb. Vasc. Biol. 2006, 26, 2523–2529.
- De Stefano, A.; Mannucci, L.; Tamburi, F.; Cardillo, C.; Schinzari, F.; Rovella, V.; Nisticò, S.; Bennardo, L.; Di Daniele, N.; Tesauro, M. Lp-PLA2, a new biomarker of vascular disorders in metabolic diseases. Int. J. Immunopathol. Pharmacol. 2019, 33, 2058738419827154.
- Nelson, T.L.; Kamineni, A.; Psaty, B.; Cushman, M.; Jenny, N.S.; Hokanson, J.; Furberg, C.; Mukamal, K.J. Lipoprotein-associated phospholipase A2 and future risk of subclinical disease and cardiovascular events in individuals with type 2 diabetes: The Cardiovascular Health Study. Diabetologia 2011, 54, 329–333.
- Sarlon-Bartoli, G.; Boudes, A.; Buffat, C.; Bartoli, M.; Piercecchi-Marti, M.; Sarlon, E.; Arnaud, L.; Bennis, Y.; Thevenin, B.; Squarcioni, C.; et al. Circulating Lipoprotein-associated Phospholipase A2 in High-grade Carotid Stenosis: A New Biomarker for Predicting Unstable Plaque. Eur. J. Vasc. Endovasc. Surg. 2012, 43, 154–159.
- Xu, D.-L.; Liu, J.-N.; Du, Y.-M.; Yao, G.-H.; Jiang, W.-D.; Wang, X.; Dong, Z.-Q.; Hao, L.; Wang, G.-Y.; Sui, S.-J.; et al. The correlation of human serum Lp-PLA2 and hs-CRP and stability of coronary atherosclerotic plaques. Zhonghua nei ke za zhi 2009, 48, 651–654.
- Khuseyinova, N.; Imhof, A.; Rothenbacher, D.; Trischler, G.; Kuelb, S.; Scharnagl, H.; Maerz, W.; Brenner, H.; Koenig, W. Association between Lp-PLA2 and coronary artery disease: Focus on its relationship with lipoproteins and markers of inflammation and hemostasis. Atherosclerosi. 2005, 182, 181–188.
- Thompson, A.; Gao, P.; Orfei, L.; Watson, S.; Di Angelantonio, E.; Kaptoge, S.; Ballantyne, C.; Cannon, C.P.; Criqui, M.; Cushman, M.; et al. Lp-PLA(2) Studies Collaboration. (2010). Lipoprotein-associated phospholipase A(2) and risk of coronary disease, stroke, and mortality: Collaborative analysis of 32 prospective studies. Lancet 2010, 375, 1536–1544.
- Vittos, O.; Toana, B.; Vittos, A.; Moldoveanu, E. Lipoprotein-associated phospholipase A2 (Lp-PLA2): A review of its role and significance as a cardiovascular biomarker. Biomarkers 2012, 17, 289–302.
- Fenning, R.S.; Burgert, M.E.; Hamamdzic, D.; Peyster, E.G.; Mohler, E.R.; Kangovi, S.; Jucker, B.M.; Lenhard, S.C.; Macphee, C.H.; Wilensky, R.L. Atherosclerotic Plaque Inflammation Varies Between Vascular Sites and Correlates With Response to Inhibition of Lipoprotein-Associated Phospholipase A 2. J. Am. Hear. Assoc. 2015, 4, e001477.
- Mendell, J.T.; Olson, E.N. MicroRNAs in Stress Signaling and Human Disease. Cell 2012, 148, 1172–1187.
- Matsuyama, H.; Suzuki, H.I. Systems and Synthetic microRNA Biology: From Biogenesis to Disease Pathogenesis. Int. J. Mol. Sci. 2019, 21, 132.
- Cipollone, F.; Felicioni, L.; Sarzani, R.; Ucchino, S.; Spigonardo, F.; Mandolini, C.; Malatesta, S.; Bucci, M.; Mammarella, C.; Santovito, D.; et al. A Unique MicroRNA Signature Associated With Plaque Instability in Humans. Stroke 2011, 42, 2556–2563.
- Soeki, T.; Yamaguchi, K.; Niki, T.; Uematsu, E.; Bando, S.; Matsuura, T.; Ise, T.; Kusunose, K.; Hotchi, J.; Tobiume, T.; et al. Plasma MicroRNA-100 Is Associated With Coronary Plaque Vulnerability. Circ. J. 2015, 79, 413–418.
- Raju, S.; Fish, J.E.; Howe, K.L. MicroRNAs as sentinels and protagonists of carotid artery thromboembolism. Clin. Sci. 2020, 134, 169–192.
- Jin, H.; Li, D.Y.; Chernogubova, E.; Sun, C.; Busch, A.; Eken, S.M.; Saliba-Gustafsson, P.; Winter, H.; Winski, G.; Raaz, U.; et al. Local Delivery of miR-21 Stabilizes Fibrous Caps in Vulnerable Atherosclerotic Lesions. Mol. Ther. 2018, 26, 1040–1055.
- He, W.; Zhu, L.; Huang, Y.; Zhang, Y.; Shen, W.; Fang, L.; Li, J.; Wang, Z.; Xie, Q. The relationship of MicroRNA-21 and plaque stability in acute coronary syndrome. Med. 2019, 98, e18049.
- Kumar, D.; Narang, R.; Sreenivas, V.; Rastogi, V.; Bhatia, J.; Saluja, D.; Srivastava, K. Circulatory miR-133b and miR-21 as Novel Biomarkers in Early Prediction and Diagnosis of Coronary Artery Disease. Genes 2020, 11, 164.
- Fazmin, I.T.; Achercouk, Z.; Edling, C.E.; Said, A.; Jeevaratnam, K. Circulating microRNA as a Biomarker for Coronary Artery Disease. Biomoleculs 2020, 10, 1354.
- Fan, X.; Wang, E.; Wang, X.; Cong, X.; Chen, X. MicroRNA-21 is a unique signature associated with coronary plaque instability in humans by regulating matrix metalloproteinase-9 via reversion-inducing cysteine-rich protein with Kazal motifs. Exp. Mol. Pathol. 2014, 96, 242–249.
- Carino, A.; De Rosa, S.; Sorrentino, S.; Polimeni, A.; Sabatino, J.; Caiazzo, G.; Torella, D.; Spaccarotella, C.; Mongiardo, A.; Strangio, A.; et al. Modulation of Circulating MicroRNAs Levels during the Switch from Clopidogrel to Ticagrelor. BioMed Res. Int. 2016, 2016, 1–5.
- Kosaka, N.; Iguchi, H.; Ochiya, T. Circulating microRNA in body fluid: A new potential biomarker for cancer diagnosis and prognosis. Cancer Sci. 2010, 101, 2087–2092.
- Izawa, H.; Amano, T. Plasma MicroRNA-100 as a Biomarker of Coronary Plaque Vulnerability. Circ. J. 2015, 79, 303–304.
- Brockdorff, N.; Ashworth, A.; Kay, G.F.; McCabe, V.M.; Norris, D.P.; Cooper, P.J.; Swift, S.; Rastan, S. The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell 1992, 71, 515–526.
- Kraczkowska, W.; Jagodziński, P.P. The Long Non-Coding RNA Landscape of Atherosclerotic Plaques. Mol. Diagn. Ther. 2019, 23, 735–749.
- Mathy, N.W.; Chen, X.-M. Long non-coding RNAs (lncRNAs) and their transcriptional control of inflammatory responses. J. Biol. Chem. 2017, 292, 12375–12382.
- Hung, J.; Scanlon, J.P.; Mahmoud, A.D.; Rodor, J.; Ballantyne, M.; Fontaine, M.A.; Temmerman, L.; Kaczynski, J.; Connor, K.L.; Bhushan, R.; et al. Novel Plaque Enriched Long Noncoding RNA in Atherosclerotic Macrophage Regulation (PELATON). Arter. Thromb. Vasc. Biol. 2020, 40, 697–713.
- Pan, Z.; Fan, Z.; Ma, J.; Liu, H.; Shen, L.; He, B.; Zhang, M. Profiling and functional characterization of circulation LncRNAs that are associated with coronary atherosclerotic plaque stability. Am. J. Transl Res 2019, 11, 3801–3815.
- Bourantas, C.V.; Garcia-Garcia, H.M.; Torii, R.; Zhang, Y.-J.; Westwood, M.; Crake, T.; Serruys, P.W. Vulnerable plaque detection: An unrealistic quest or a feasible objective with a clinical value? Hear. 2016, 102, 581–589.
- Shimamura, K.; Kubo, T.; Akasaka, T. Evaluation of coronary plaques and atherosclerosis using optical coherence tomography. Expert Rev. Cardiovasc. Ther. 2021, 19, 379–386.
- Su, M.-I.; Chen, C.-Y.; Yeh, H.-I.; Wang, K.-T. Concise Review of Optical Coherence Tomography in Clinical Practice. Acta Cardiol. Sin. 2016, 32, 381–386.
- Lv, R.; Maehara, A.; Matsumura, M.; Wang, L.; Wang, Q.; Zhang, C.; Guo, X.; Samady, H.; Giddens, D.P.; Zheng, J.; et al. Using optical coherence tomography and intravascular ultrasound imaging to quantify coronary plaque cap thickness and vulnerability: A pilot study. Biomed. Eng. Online 2020, 19, 1–18.
- Bourantas, C.V.; Jaffer, F.A.; Gijsen, F.J.; van Soest, G.; Madden, S.P.; Courtney, B.K.; Fard, A.M.; Tenekecioglu, E.; Zeng, Y.; Van Der Steen, A.F.; et al. Hybrid intravascular imaging: Recent advances, technical considerations, and current applications in the study of plaque pathophysiology. Eur. Hear. J. 2017, 38, 400–412.
- Ali, Z.; Landmesser, U.; Galougahi, K.K.; Maehara, A.; Matsumura, M.; Shlofmitz, R.A.; Guagliumi, G.; Price, M.J.; Hill, J.M.; Akasaka, T.; et al. Optical coherence tomography-guided coronary stent implantation compared to angiography: A multicentre randomised trial in PCI – design and rationale of ILUMIEN IV: OPTIMAL PCI. EuroIntervention 2021, 16, 1092–1099.
- Gardner, C.M.; Tan, H.; Hull, E.L.; Lisauskas, J.B.; Sum, S.T.; Meese, T.M.; Jiang, C.; Madden, S.P.; Caplan, J.D.; Burke, A.P.; et al. Detection of Lipid Core Coronary Plaques in Autopsy Specimens With a Novel Catheter-Based Near-Infrared Spectroscopy System. JACC: Cardiovasc. Imaging 2008, 1, 638–648.
- Fard, A.M.; Vacas-Jacques, P.; Hamidi, E.; Wang, H.; Carruth, R.W.; Gardecki, J.A.; Tearney, G.J. Optical coherence tomography – near infrared spectroscopy system and catheter for intravascular imaging. Opt. Express 2013, 21, 30849–30858.

