 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Laia Sadeghi | + 3302 word(s) | 3302 | 2021-06-16 11:47:03 | | | |
| 2 | Camila Xu | + 430 word(s) | 3732 | 2021-06-28 05:39:56 | | | | |
| 3 | Camila Xu | -40 word(s) | 3692 | 2021-06-28 05:41:19 | | | | |
| 4 | Camila Xu | -40 word(s) | 3692 | 2021-06-28 05:41:53 | | |
Video Upload Options
Lymphocyte migration to and sequestration in specific microenvironments plays a crucial role in their differentiation and survival. Lymphocyte trafficking and homing is tightly regulated by signaling pathways and is mediated by cytokines, chemokines, cytokine/chemokine receptors, and adhesion molecules. The production of cytokines and chemokines is largely controlled by transcription factors in the context of a specific epigenetic landscape. These regulatory factors are strongly inter-connected, and they influence the gene expression pattern in lymphocytes, promoting processes such as cell survival. The epigenetic status of the genome plays a key role in regulating gene expression during many key biological processes and it is becoming more evident that dysregulation of epigenetic mechanisms contributes to cancer initiation, progression and drug resistance.
1. Epigenetic Regulation of Tumor-Microenvironment
The tumor microenvironment, which includes various types of cells, is a location where tumor cells continuously interact with surrounding normal cells and extracellular molecules. In B-cell lymphomas, interaction of tumor cells with stromal cells in the tumor microenvironment promotes tumor cell growth, proliferation, survival and drug resistance [1][2][3]. The interactions between malignant B-cells and tumor microenvironment components alter the gene expression profiles of tumor cells in order to favor the survival of malignant B-cells [4][5][6].
Mantle cell lymphoma (MCL) is a rare and largely incurable B-cell non-Hodgkin lymphoma (NHL) accounting for 5–10% of all lymphomas [7]. This results in cyclin D1 overexpression and leads to aberrant cell cycle progression [8][9]. These studies demonstrated several recurrent mutations, includingATM,TP53andMLL2,but they also uncovered a number of novel and infrequent mutations in patients, which suggests divergent clonal evolution pattern in MCL cells [10]. As tumor cells divide, mutations occur randomly, resulting in multiple sub-clonal populations with different phenotypes within the same tumor, known as intra-tumor heterogeneity, which makes tumor more prone to recurrence and drug resistance.
Distinct from mutations, altered epigenetic status, i.e., chromosomal conditions that influence gene expression without changing the primary DNA sequence, plays an important role in the development of B-cell lymphomas [11][12][13]. Variable epigenetic components throughout the genome that influence gene expression include DNA methylation and patterns of histone modification, which alter chromatin structure, DNA accessibility and gene expression patterns [14]. The combination of genetic mutations and epigenetic changes induced by internal and external factors leads to constitutive activation of proto-oncogenes and the loss of tumor-suppressor gene activity that ultimately causes tumorigenesis [11][15][16]. Comprehensive methylation profiling by genome-wide array, comparing DNA methylation changes in MCL patients with normal B-cells, has demonstrated highly heterogenous methylation profiles across MCL patient samples [17].
In addition to the role of intrinsic abnormalities in tumor cells, an essential role of extrinsic factors, such as the tumor microenvironment, in tumor growth and development of drug resistance in B-cell lymphomas is widely accepted [18][19][20]. Interactions between tumor cells and microenvironment components trigger activation of a cascade of signals that travel to the nucleus, integrate with the specific epigenetic landscape of the tumor cell and thereafter cause specific alterations in gene expression. Crosstalk between tumor cells and microenvironment components promotes anti-tumor immune responses in B-cell lymphomas [21]. Although the role of the tumor microenvironment in the regulation of cellular transduction pathways in tumor cells has already been described [6][22][23], much less is known about the interaction between cell signaling events and the specific epigenetic landscape of the cells that causes cell-type-specific effects on chromatin structure and thereby gene expression.
Here, we review current evidence indicating that epigenetic alterations contribute to the development of NHLs including MCL. Recent investigations have identified mutations in epigenetic regulators that cause epigenetic changes and thereby lead to abnormal gene activity in lymphocytes [16][24][25]. However, epigenetic alterations in lymphocytes have also been observed in the absence of mutations in genes encoding proteins important for epigenetic regulation, suggesting that the tumor microenvironment can induce epigenetic dysregulation in lymphoma cells. We further propose a possible model where somatic mutations and the tumor microenvironment interact in order to shape the epigenetic landscape of lymphoma cells, which induces transcriptional changes in tumor cells, enabling their proliferation and survival.
2. Epigenetic Alteration in MCL
Chromatin structure and organization play a crucial role in regulating transcription by modulating DNA accessibility to the transcriptional machinery and transcription factors. The establishment and maintenance of distinct chromatin domains play an important role by establishing the pre-conditions that determine the extent to which individual genes can be regulated by transcription factors [26]. Multiple mechanisms affecting chromatin organization, including DNA methylation, ATP-dependent chromatin remodeling and post-translational modification of histones, are required to ensure proper cellular functions. Different modifications of histone proteins are associated with discrete chromatin states that may be activated or repressed and impact a wide variety of cellular processes including transcription, DNA replication and DNA repair [27].
Currently known disease-causing epigenetic changes in B-cell lymphomas are predominantly induced by somatic mutations in epigenetic regulators, which have been identified by genome-wide screens and include key players involved in DNA methylation, histone modification and chromatin organization [28][25][29][30]. Here, we will briefly discuss mutations that modulate the epigenetic landscape of MCL as well as some other lymphomas, resulting in an abnormal transcriptional programming (Figure 1). A list of mutations known to be involved in epigenetic alterations in MCL is presented in Table 1.
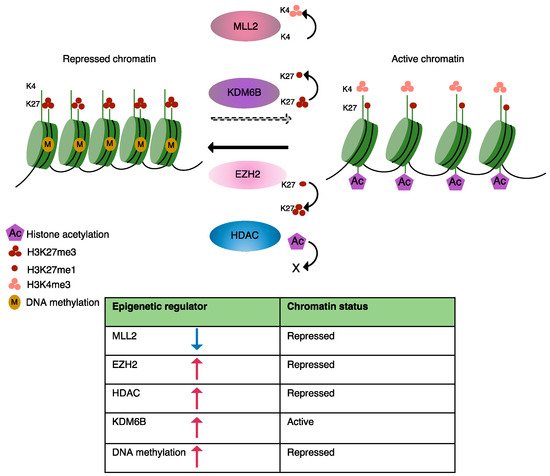
| Dysregulated Epigenetic Mark in MCL | Gene/Genes Mediating Epigenetic Dysregulation in MCL | Type of Dysregulation | Target Gene/Genes Affected by Epigenetic Dysregulation | Putative Role in MCL | Reference |
|---|---|---|---|---|---|
| DNA methylation | - | DNA hypomethylation at promoter region of target gens | Mediating the expression of SOX11 | Promotes oncogenic cell proliferation | [31] |
| DNA methylation | DNMT1 upregulation | Global DNA hypermethylation | Abnormal expression of β-catenin which upregulates the expression of c-MYC and MMP7 (Matrix Metallopeptidase 7) Reduced expression of tumor suppressor gene; PARG1 |
Promotes tumor cell proliferation and survival | [32][33] |
| Histone H3K27acetylation | Mutation in the component of SWI-SNF complex (SMARCA4) | Abnormal histone H3K27 acetylation and chromatin accessibility at the promoter/enhancer region of target genes | Reduced chromatin accessibility at promoter region of transcription factor ATF3 (negative regulator of anti-apoptotic gene BCL-xL) | MCL cell survival and drug resistance | [28][34][35] |
| Global histone acetylation | Abnormal activity of HDACs, Class I, II, e.g., HDAC8 | Enhanced HDAC (Histone deacetylase) activity leading to abnormal histone acetylation and chromatin accessibility | Reduced transcription of pro-apoptotic genes (BIM, BMF) Enhanced expression of c-MYC and PLK1 |
Inhibits apoptosis and promotes tumor cell proliferation and survival | [36][37][38][39] |
| Histone H3K36me3 | Gain of function mutation in histone methyltransferase WHSC1 (MMSET) | Enhanced H3K36me3 levels | Enhanced expression of cell cycle regulators | Promotes tumor cell proliferation | [40][41] |
| Histone H3K4 methylation | Loss of function mutation in histone methyltransferase MLL2 (KMT2D) | Diminished H3K4 methylation levels | Functional consequences in MCL are not well understood Contributes to genome instability and transcriptional stress |
Disturbs the expression of genes that sustain proliferation and cell survival | [28][42][43][44] |
| H3K27me3 | EZH2 upregulation | Enhanced H3K27me3 levels | Repressed expression of CDKN2B, HOX genes | Promotes MCL cell growth | [45][46] |
| H3K27me3 | KDM6B histone demethylase | Enhanced KDM6B levels, reduces H3K27me3 at promoter region of target genes | Functional consequences in MCL are not well understood In other B-cell malignancies target NF-κB subunits and target genes |
Promoters tumor cells survival and drug resistance | [47][48][49] |
Extensive perturbation of cytosine methylation patterns is a hallmark of B-cell malignancies and occurs in specific patterns that can be distinguished in different subtypes of NHLs [31]. Analysis of the genome-wide DNA methylation profile in MCL patients revealed significantly different promoter methylation patterns between MCL patient samples and normal B-cells [31]. who showed that DNA methyltransferase-1 (DNMT-1) is upregulated in MCL, causing global disruption of DNA methylation patterns [32]. In general, abnormal DNA methylation of tumor suppressor genes suppresses their transcription levels, and this is a common phenomenon observed in cancer.
Nucleosomes play an important role in regulating higher-order chromatin structure. Chromatin remodeling enzymes use ATP to alter the structure and/or position of nucleosomes and thereby regulate the accessibility of DNA to transcription factors [50]. In eukaryotic cells, a wide variety of chromatin-remodeling complexes are present to remodel chromatin via different mechanisms [51]. For example, the SWI/SNF family of chromatin remodeling enzymes globally moves or evicts nucleosomes in order to create open chromatin regions, leading to transcriptional activation of genes throughout the genome [52].
Mutations in genes encoding proteins involved in chromatin remodeling have been identified in different types of cancer. SMARCA4, a central component of the SWI/SNF chromatin-remodeling complex, regulates transcription via remodeling nucleosome positions. MCL patients with aSMARCA4mutation are resistant to combined therapy with Ibrutinib (BTK inhibitor) and Venetoclax (BCL-2 inhibitor) The mutation inSMARCA4affects chromatin accessibility and reduces expression of the transcription factor, ATF3, which negatively regulates transcription of the anti-apoptotic geneBCL-xL,
In general, histone acetylation is associated with active transcription. CBP, known as (P300/CBP), acetylates histone and non-histone proteins in order to activate transcription [53]. In contrast, histone deacetylases (HDACs) remove acetyl groups from hyperacetylated histones in order to suppress gene transcription [54]. In addition to histone deacetylation, HDACs also regulate the acetylation status of a variety of non-histone proteins [55].
HDAC inhibitors (HDACi) are a class of chemical compounds that increase histone acetylation and restore the balance between pro- and anti-apoptotic proteins and induce apoptosis in tumor cells. Inhibition of HDACs induced cell death in MCL cell lines by enhancing histone acetylation at the promoter region of pro-apoptotic genes including (BIMandBMF), which induced their transcription [36]. In addition, Vorinostat in combination with Palbociclib (selective inhibitor of cyclin-dependent kinase 4/6 involved in cell cycle progression) reduced MCL cell growth and induced apoptosis significantly by increasing histone H3 acetylation and inhibiting BCL-2 family members. Overexpression of BCL-2 family proteins attenuates apoptosis in tumor cells [37].
Another HDACi (PCI-24781) enhanced acetylation of histone H3 at the promoter region of the gene encoding P21 (CDKN1A), causing increased chromatin accessibility for binding of transcription factors [38]. Moreover, PCI-24781 significantly downregulated the expression of NF-κB subunits (NF-κB1 and REL B) and NF-κB This suggests a role for histone deacetylation in the regulation of NF-κB activity, which remains poorly understood. A combination of PCI-24781 and bortezomib (a proteasome inhibitor) synergistically suppressed NF-κB and induced apoptosis in lymphoma cells [38].
HDAC3 has been implicated as a regulator of PD-L1 expression in B-cell lymphomas [56]. Inhibition of HDAC3 reversed microenvironment-mediated immunosuppression and resulted in better clinical response to PD-L1 blockage [56]. In Chronic lymphocytic leukemia (CLL), the combination of HDAC6 inhibitor (ACY-738) with PD-L1 blockage using monoclonal antibodies significantly increased anti-tumor efficiency of T-cells and reduced tumor burden [57]. Thus, the combination of HDAC inhibitors and checkpoint blockage can help to overcome immunotherapy resistance.
Post-translational modification of histone proteins by methylation is an important and common modification that influences many cellular processes. The major methylation sites on histone proteins are lysine and arginine residues and methylation of non-histone proteins is also frequently observed [58][59]. Histone methylation is a dynamic process, and methyl groups can be removed from histone proteins by histone demethylases [60]. The abnormal expression of various methyltransferases and demethylases has been reported in many tumor types, suggesting that the level and degree of histone methylation contributes to tumorigenesis and that the enzymes involved provide promising targets for anti-cancer treatment [47][61][62].
WHSC1 (also known as MMSET or NSD2) is a specific histone methyltransferase, responsible for histone H3 methylation at lysine 36, and it is mutated in a subgroup of B-cell malignancies including MCL [40][41]. Alterations in histone methylation status can switch a gene from an active to a repressed state or vice versa. As a result of altered WHSC1 levels, the expression of genes positively involved in proliferation and cell-cycle progression increases, suggesting thatWHSC1functions as an oncogene favoring tumor cell growth and proliferation.
While H3K4me3 is associated with the activation of gene expression, H3K27me3 is a mark for repressed genes [63][64]. The promoter region is generally uniquely marked with either H3K4me3 or H3K27me3, except in the case of bivalent chromatin at the promoter region of developmental regulatory genes [63][64][65]. Bivalent chromatin is marked with histone modifications associated with both gene activation and repression and keeps repressed genes poised and ready for rapid activation [65]. Bernhart S et al., using ChIP-seq (chromatin immunoprecipitation combined with high-throughput sequencing), demonstrated that chromatin structure at bivalent promoters in cancer cells (both in solid and hematological tumors) is disrupted, resulting in deregulation of gene silencing and activation of poised genes [66].
MLL2has been identified as one of the commonly mutated genes in NHLs including Diffuse large B-cell lymphoma (DLBCL) and MCL [34][35][42]. In DLBCL, mutation ofMLL2produces a truncatedMLL2protein that lacks the SET domain (required for its methyltransferase activity) [42]. Although the role ofMLL2in MCL progression is not well understood,MLL2loss-of-function mutations are known to diminish H3K4 methylation and drive tumor cell growth and proliferation, suggesting thatMLL2functions as a tumor suppressor gene [43][67]. Interestingly, a recent study has provided evidence for the role ofMLL2in maintaining genomic stability, since the loss ofMLL2caused chromosomal aberrations and accumulation of mutations [44].
The multi-protein polycomb complexes (PRCs) promote transcriptional repression by disturbing nucleosome occupancy and mediating H3K27 methylation. All three forms of H3K27 methylation are mutually exclusive and are present at distinct genomic regions. Trimethylated H3K27 (H3K27me3) is present at promoter regions of silenced genes and is associated with transcriptional repression [68]. To preserve chromatin structure and prolong gene silencing, H3K27me3 recruits PRC complexes to nucleosomes of the nascent DNA strand during DNA replication in order to maintain H3K27me3 levels [69].
One of the most frequent genetic alterations observed in germinal center B-cell (GCB) lymphomas such as Follicular lymphoma (FL) and DLBCL is mutations ofEZH2that affect the SET domain. EZH2is essential for normal B-cell development and rearrangement of the immunoglobulin heavy chain (IGHV) [70]. Moreover,EZH2is required for the differentiation of germinal B-cells into memory cells through the establishment and maintenance of bivalent modifications at the promoter region of key regulatory genes [71].
In addition to mutation, other genetic lesions affectingEZH2, including chromosomal gain or loss and DNA hypermethylation, have been identified in B-cell malignancies The work of Demosthenous et al. showed that deregulation of PRC2/EZH2 in MCL cell lines mediates epigenetic silencing of theCDKN2BCDK inhibitor gene to promote MCL cell proliferation and survival [45]. In addition, it has been shown that EZH2 overexpression in MCL cell lines and primary cells facilitates recruitment of the DNA methylation machinery, enabling more stable and long-term repression of HOX genes [46]. Further studies are required to develop strategies to specifically alter H3K27me3 levels in tumor cells.
Post-translational modification of histone H3 at lysine 27 has an essential role in both transcriptional repression and activation. Studies have shown that both increase and decrease in the activity of enzymes regulating H3K27me3 levels are associated with carcinogenesis [29][48]. Despite the opposing activities of EZH2 and the KDM6B demethylase in affecting H3K27me3 levels, both these enzymes have been implicated in both lymphoma development and drug resistance.
Histone demethylase JMJD3 removes di- and tri-methyl marks from H3K27 and reverses PRC2/EZH2-mediated transcription repression [72][73]. In contrast, KDM6B overexpression has been observed in Hodgkin’s lymphomas (HLs), DLBCL, Multiple myeloma (MM) and Acute myeloid leukemia (AML) KDM6B is required for maintaining hematopoietic stem cell (HSC) self-renewal ability and repopulation capacity and plays an important role in the differentiation of memory B-cells [74][75].
In MM, KDM6B regulates the expression of key components of the MAP Kinase pathway in a demethylase-activity-independent manner in order to enhance tumor cell survival, whereas in DLBCL, KDM6B controls BCL6 expression and protein levels in a fashion that requires the demethylase activity in order to facilitate tumor cell proliferation [47][48]. KDM6B has been identified as a new therapeutic target in B-cell malignancies, and its selective inhibitor GSK-J4 has been shown to counteract cell proliferation in different tumor types such as AML, MM and DLBCL [48][76].
KDM6B overexpression can antagonize EZH2-mediated repression [77] of tumor suppressor genes in solid tumors by reducing H3K27me3 levels, while inhibition of KDM6B boosts apoptotic response to PI3K/AKT inhibitor treatment. This suggests that KDM6B can act as a tumor suppressor gene or an oncogene depending on cellular context [78].
3. Tumor-Microenvironment Mediated Epigenetic Alterations
As discussed above, cancer-associated mutations that affect the epigenetic machinery can lead to imbalances that change the epigenetic landscape in cancer cells and its effect on gene expression programs. Regulation of gene expression in response to signaling pathways requires post-translational modifications of histones and nucleosome rearrangement, which promotes chromatin reorganization, leading to activation or repression of transcription at particular loci throughout the genome. Interactions between malignant B-cells and stromal cells activate multiple signaling pathways, including PI3K/AKT and NF-κB, that function as signal transducers for cell-surface receptors, particularly receptor tyrosine kinases, in order to alter cellular function [6]. Here, we focus on some examples of key tumor microenvironment-mediated signaling pathways that have a direct impact on the chromatin state of tumor cells.
The PI3K/AKT pathway plays a key role in many cellular mechanisms and is often dysregulated in malignant B-cells [79]. The PI3K/AKT pathway regulates essential cellular functions including growth, proliferation, metabolism, differentiation and motility in normal and malignant B-cells [80]. to generate phosphatidylinositol-3,4,5-trisphosphate (PIP3), which in turn acts as a second messenger to assemble and activate downstream signaling complexes, including protein kinase B, also known as AKT [81]. Phosphorylated AKT mediates phosphorylation of different proteins, including the mammalian target of rapamycin (mTOR), and the IκB inhibitor of NF
Adhesion of MCL cells to stromal cells in the tumor microenvironment activates the PI3K pathway, which transmits signals from cell membrane receptors, such as the B-cell receptor and CXCR4 (chemokine receptor), to the nucleus, leading to transcriptional regulation of genes involved in migration, proliferation and survival [82][79][83][84]. AKT-mediated phosphorylation of P300/CBP facilitates recruitment of the transcription machinery at the promoter region of genes involved in the survival of MCL and CLL cells [79][85]. Expression levels of CXCR4 are regulated by the transcription factor, FOXO1, and acetylation of histones by P300/CBP is a prerequisite for FOXO1/P300-mediated recruitment of transcription machinery at the promoter regions of important target genes [86][87]. Zhou XR et al. reported that a P300/CBP inhibitor (A-485) overcomes resistance to Idelalisib in MCL by reducing histone acetylation at the promoter region of receptor tyrosine kinase (RTK) genes, thereby, reducing their transcriptional upregulation by an independent mechanism [88].
In mammals, DNA methylation is performed by DNMT1 and DNMT3, which are functionally and structurally distinct [89]. In general, maintenance of DNA methylation activity is essential for the preservation of genomic integrity and inhibition of inappropriate gene activation. Abnormal activation of PI3K/AKT might contribute to tumor cell growth by favoring DNA hypermethylation and chromatin compaction at the promoter region of specific target genes, thereby suppressing their expression [90]. Accumulating evidence demonstrates a role for DNMT1 in malignant progression of NHLs including DLBCL and MCL by enhancing cell cycle progression [32][91][92].
In addition, PI3K/AKT is involved in regulating histone methylation levels. AKT interacts with and phosphorylates EZH2 at Serine 21 and suppresses its methyltransferase activity, which results in reduced H3K27me3 levels at the promoter regions of target genes, thus disrupting gene silencing [93]. A Serine to Alanine substitution at the phosphorylation site increases EZH2 association with H3 [93].
Tumor microenvironment-mediated upregulation of EZH2 has been reported in various types of NHL [94][95]. In a subset of CLL cells, signals from the microenvironment result in pronounced upregulation of EZH2 mRNA and protein levels, which induces anti-apoptotic responses and enhances cell viability [94]. Moreover, dysregulation of EZH2 levels in CLL results in intra-tumor heterogeneity contributing to drug resistance [96].
NF-κB binds to the promoter and/or enhancer region of its target genes, which are involved in stress responses, cell proliferation and apoptosis, and regulates their transcription [97]. The NF–κB The NF-κB signaling pathway is divided into separate canonical and non-canonical pathways that regulate distinct gene expression programs. Disruption of the NF-κB
Activation of NF-κB in response to cytokines and chemokines in tumor microenvironments promotes upregulation of EZH2 in leukemia and lymphoma cell lines [98]. Natoli G et al. showed that exposure of macrophages to different inflammatory cytokines elevated KDM6B In a recent study from our lab, we showed that stromal cell adhesion mediates overexpression of KDM6B in MCL cell lines, which could potentially lead to the removal of the H3K27me3 mark from the promoter regions of genes encoding NF-κB subunits and thus activate their expression and nuclear localization. activity mediates transcription of migration and adhesion-related genes and promotes tumor cell survival.
4. Targeting PI3K in MCL
Given the crucial role of PI3K/AKT in lymphoma development and survival, different compounds have been identified that inhibit the PI3K activity. Idelalisib (formerly known as GS-1101 and CAL-101), the first class of PI3K inhibitors, has been used as a single-agent monotherapy or in combination with other inhibitors/agents to treat refractory or relapsed MCL and CLL patients [99]. Idelalisib inhibits MCL and CLL cell migration toward CXCL12 gradients, promotes egression of malignant B-cells from lymph nodes and induces apoptosis of malignant B-cells [99][100][101].
Combined PI3K and HDAC inhibitors have demonstrated significant anti-tumor effects in MCL and DLBCL [39][102]. CUDC-907 (dual PI3K and HDAC inhibitor) inhibits growth and proliferation of both Ibrutinib-sensitive and Ibrutinib-resistant MCL primary cells in vitro and induces apoptosis and cell cycle arrest through downregulation of PLK1, polo-like kinase 1, a key regulator of mitosis [39]. Moreover, CUDC-907 downregulated MYC in DLBCL and showed a promising anti-tumor effect [102]. Given the role of PI3K/AKT in priming chromatin for transcriptional activation, inhibiting its kinase activity can cause transcriptional repression of oncogenes, such as MYC [84].
References
- Sadeghi, L.; Arvidsson, G.; Merrien, M.; Wasik, A.M.; Görgens, A.; Smith, C.E.; Sander, B.; Wright, A.P. Differential B-Cell Receptor Signaling Requirement for Adhesion of Mantle Cell Lymphoma Cells to Stromal Cells. Cancers 2020, 12, 1143.
- Lenz, G.; Wright, G.; Dave, S.; Xiao, W.; Powell, J.; Zhao, H.; Xu, W.; Tan, B.; Goldschmidt, N.; Iqbal, J.; et al. Stromal Gene Signatures in Large-B-Cell Lymphomas. N. Engl. J. Med. 2008, 359, 2313–2323.
- Burger, J.A.; Tsukada, N.; Burger, M.; Zvaifler, N.J.; Dell’Aquila, M.; Kipps, T.J. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood 2000, 96, 2655–2663.
- Sonugur, F.G.; Akbulut, H. The Role of Tumor Microenvironment in Genomic Instability of Malignant Tumors. Front. Genet 2019, 10, 1063.
- Arvidsson, G.; Henriksson, J.; Sander, B.; Wright, A.P. Mixed-species RNAseq analysis of human lymphoma cells adhering to mouse stromal cells identifies a core gene set that is also differentially expressed in the lymph node microenvironment of mantle cell lymphoma and chronic lymphocytic leukemia patients. Haematologica 2018, 103, 666–678.
- Herishanu, Y.; Perez-Galan, P.; Liu, D.; Biancotto, A.; Pittaluga, S.; Vire, B.; Gibellini, F.; Njuguna, N.; Lee, E.; Stennett, L.; et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood 2011, 117, 563–574.
- Ghielmini, M.; Zucca, E. How I treat mantle cell lymphoma. Blood 2009, 114, 1469–1476.
- Bosch, F.; Jares, P.; Campo, E.; Lopez-Guillermo, A.; Piris, M.A.; Villamor, N.; Tassies, D.; Jaffe, E.S.; Montserrat, E.; Rozman, C.; et al. PRAD-1/cyclin D1 gene overexpression in chronic lymphoproliferative disorders: A highly specific marker of mantle cell lymphoma. Blood 1994, 84, 2726–2732.
- Raffeld, M.; Jaffe, E.S. bcl-1, t(11;14), and mantle cell-derived lymphomas. Blood 1991, 78, 259–263.
- Wu, C.; de Miranda, N.F.; Chen, L.; Wasik, A.M.; Mansouri, L.; Jurczak, W.; Galazka, K.; Dlugosz-Danecka, M.; Machaczka, M.; Zhang, H.; et al. Genetic heterogeneity in primary and relapsed mantle cell lymphomas: Impact of recurrent CARD11 mutations. Oncotarget 2016, 7, 38180–38190.
- Di Pietro, A.; Good-Jacobson, K.L. Disrupting the Code: Epigenetic Dysregulation of Lymphocyte Function during Infectious Disease and Lymphoma Development. J. Immunol. 2018, 201, 1109–1118.
- Yusufova, N.; Kloetgen, A.; Teater, M.; Osunsade, A.; Camarillo, J.M.; Chin, C.R.; Doane, A.S.; Venters, B.J.; Portillo-Ledesma, S.; Conway, J.; et al. Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture. Nature 2021, 589, 299–305.
- Darwiche, N. Epigenetic mechanisms and the hallmarks of cancer: An intimate affair. Am. J. Cancer Res. 2020, 10, 1954–1978.
- Feinberg, A.P.; Koldobskiy, M.A.; Gondor, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299.
- Chatterjee, A.; Rodger, E.J.; Eccles, M.R. Epigenetic drivers of tumourigenesis and cancer metastasis. Semin. Cancer Biol. 2018, 51, 149–159.
- Shaknovich, R.; Melnick, A. Epigenetics and B-cell lymphoma. Curr. Opin. Hematol. 2011, 18, 293–299.
- Enjuanes, A.; Albero, R.; Clot, G.; Navarro, A.; Bea, S.; Pinyol, M.; Martin-Subero, J.I.; Klapper, W.; Staudt, L.M.; Jaffe, E.S.; et al. Genome-wide methylation analyses identify a subset of mantle cell lymphoma with a high number of methylated CpGs and aggressive clinicopathological features. Int. J. Cancer 2013, 133, 2852–2863.
- Zhang, L.; Yang, J.; Qian, J.; Li, H.; Romaguera, J.E.; Kwak, L.W.; Wang, M.; Yi, Q. Role of the microenvironment in mantle cell lymphoma: IL-6 is an important survival factor for the tumor cells. Blood 2012, 120, 3783–3792.
- Shain, K.H.; Dalton, W.S.; Tao, J. The tumor microenvironment shapes hallmarks of mature B-cell malignancies. Oncogene 2015, 34, 4673–4682.
- Burger, J.A.; Ghia, P.; Rosenwald, A.; Caligaris-Cappio, F. The microenvironment in mature B-cell malignancies: A target for new treatment strategies. Blood 2009, 114, 3367–3375.
- Armengol, M.; Santos, J.C.; Fernandez-Serrano, M.; Profitos-Peleja, N.; Ribeiro, M.L.; Roue, G. Immune-Checkpoint Inhibitors in B-Cell Lymphoma. Cancers 2021, 13, 214.
- Medina, D.J.; Goodell, L.; Glod, J.; Gelinas, C.; Rabson, A.B.; Strair, R.K. Mesenchymal stromal cells protect mantle cell lymphoma cells from spontaneous and drug-induced apoptosis through secretion of B-cell activating factor and activation of the canonical and non-canonical nuclear factor kappa B pathways. Biomarker Res. 2012, 97, 1255–1263.
- Saba, N.S.; Liu, D.; Herman, S.E.; Underbayev, C.; Tian, X.; Behrend, D.; Weniger, M.A.; Skarzynski, M.; Gyamfi, J.; Fontan, L.; et al. Pathogenic role of B-cell receptor signaling and canonical NF-kappaB activation in mantle cell lymphoma. Blood 2016, 128, 82–92.
- Wang, C.Y.; Tian, C.; Zhang, Y.Z. [Research Advances in the Epigenetics of Diffuse Large B-Cell Lymphoma-Review]. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2017, 25, 1232–1236.
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303.
- Filion, G.J.; van Bemmel, J.G.; Braunschweig, U.; Talhout, W.; Kind, J.; Ward, L.D.; Brugman, W.; de Castro, I.J.; Kerkhoven, R.M.; Bussemaker, H.J.; et al. Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell 2010, 143, 212–224.
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638.
- Bea, S.; Valdes-Mas, R.; Navarro, A.; Salaverria, I.; Martin-Garcia, D.; Jares, P.; Gine, E.; Pinyol, M.; Royo, C.; Nadeu, F.; et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 18250–18255.
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185.
- Argatoff, L.H.; Connors, J.M.; Klasa, R.J.; Horsman, D.E.; Gascoyne, R.D. Mantle cell lymphoma: A clinicopathologic study of 80 cases. Blood 1997, 89, 2067–2078.
- Queiros, A.C.; Beekman, R.; Vilarrasa-Blasi, R.; Duran-Ferrer, M.; Clot, G.; Merkel, A.; Raineri, E.; Russinol, N.; Castellano, G.; Bea, S.; et al. Decoding the DNA Methylome of Mantle Cell Lymphoma in the Light of the Entire B Cell Lineage. Cancer Cell 2016, 30, 806–821.
- Li, X.Y.; Li, Y.; Zhang, L.; Liu, X.; Feng, L.; Wang, X. The antitumor effects of arsenic trioxide in mantle cell lymphoma via targeting Wnt/betacatenin pathway and DNA methyltransferase-1. Oncol. Rep. 2017, 38, 3114–3120.
- Ripperger, T.; von Neuhoff, N.; Kamphues, K.; Emura, M.; Lehmann, U.; Tauscher, M.; Schraders, M.; Groenen, P.; Skawran, B.; Rudolph, C.; et al. Promoter methylation of PARG1, a novel candidate tumor suppressor gene in mantle-cell lymphomas. Haematologica 2007, 92, 460–468.
- Zhang, J.; Jima, D.; Moffitt, A.B.; Liu, Q.; Czader, M.; Hsi, E.D.; Fedoriw, Y.; Dunphy, C.H.; Richards, K.L.; Gill, J.I.; et al. The genomic landscape of mantle cell lymphoma is related to the epigenetically determined chromatin state of normal B cells. Blood 2014, 123, 2988–2996.
- Agarwal, R.; Chan, Y.C.; Tam, C.S.; Hunter, T.; Vassiliadis, D.; Teh, C.E.; Thijssen, R.; Yeh, P.; Wong, S.Q.; Ftouni, S.; et al. Dynamic molecular monitoring reveals that SWI-SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat. Med. 2019, 25, 119–129.
- Xargay-Torrent, S.; Lopez-Guerra, M.; Saborit-Villarroya, I.; Rosich, L.; Campo, E.; Roue, G.; Colomer, D. Vorinostat-induced apoptosis in mantle cell lymphoma is mediated by acetylation of proapoptotic BH3-only gene promoters. Clin. Cancer Res. 2011, 17, 3956–3968.
- Chaturvedi, N.K.; Hatch, N.D.; Sutton, G.L.; Kling, M.; Vose, J.M.; Joshi, S.S. A novel approach to eliminate therapy-resistant mantle cell lymphoma: Synergistic effects of Vorinostat with Palbociclib. Leuk. Lymphoma 2019, 60, 1214–1223.
- Bhalla, S.; Balasubramanian, S.; David, K.; Sirisawad, M.; Buggy, J.; Mauro, L.; Prachand, S.; Miller, R.; Gordon, L.I.; Evens, A.M. PCI-24781 Induces Caspase and Reactive Oxygen Species-Dependent Apoptosis Through NF-kappa B Mechanisms and Is Synergistic with Bortezomib in Lymphoma Cells. Clin. Cancer Res. 2009, 15, 3354–3365.
- Guo, H.; Zeng, D.; Zhang, H.; Bell, T.; Yao, J.; Liu, Y.; Huang, S.; Li, C.J.; Lorence, E.; Zhou, S.; et al. Dual inhibition of PI3K signaling and histone deacetylation halts proliferation and induces lethality in mantle cell lymphoma. Oncogene 2019, 38, 1802–1814.
- Yang, P.; Zhang, W.; Wang, J.; Liu, Y.; An, R.; Jing, H. Genomic landscape and prognostic analysis of mantle cell lymphoma. Cancer Gene Ther. 2018, 25, 129–140.
- Oyer, J.A.; Huang, X.; Zheng, Y.; Shim, J.; Ezponda, T.; Carpenter, Z.; Allegretta, M.; Okot-Kotber, C.I.; Patel, J.P.; Melnick, A.; et al. Point mutation E1099K in MMSET/NSD2 enhances its methyltranferase activity and leads to altered global chromatin methylation in lymphoid malignancies. Leukemia 2014, 28, 198–201.
- Pasqualucci, L. The genetic basis of diffuse large B-cell lymphoma. Curr. Opin. Hematol. 2013, 20, 336–344.
- Zhang, J.; Dominguez-Sola, D.; Hussein, S.; Lee, J.E.; Holmes, A.B.; Bansal, M.; Vlasevska, S.; Mo, T.; Tang, H.; Basso, K.; et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat. Med. 2015, 21, 1190–1198.
- Kantidakis, T.; Saponaro, M.; Mitter, R.; Horswell, S.; Kranz, A.; Boeing, S.; Aygun, O.; Kelly, G.P.; Matthews, N.; Stewart, A.; et al. Mutation of cancer driver MLL2 results in transcription stress and genome instability. Genes Dev. 2016, 30, 408–420.
- Demosthenous, C.; Gupta, S.K.; Sun, J.; Wang, Y.S.; Troska, T.P.; Gupta, M. Deregulation of Polycomb Repressive Complex-2 in Mantle Cell Lymphoma Confers Growth Advantage by Epigenetic Suppression ofcdkn2b. Front. Oncol. 2020, 10.
- Kanduri, M.; Sander, B.; Ntoufa, S.; Papakonstantinou, N.; Sutton, L.A.; Stamatopoulos, K.; Kanduri, C.; Rosenquist, R. A key role for EZH2 in epigenetic silencing of HOX genes in mantle cell lymphoma. Epigenetics 2013, 8, 1280–1288.
- Mathur, R.; Sehgal, L.; Havranek, O.; Kohrer, S.; Khashab, T.; Jain, N.; Burger, J.A.; Neelapu, S.S.; Davis, R.E.; Samaniego, F. Inhibition of demethylase KDM6B sensitizes diffuse large B-cell lymphoma to chemotherapeutic drugs. Haematologica 2017, 102, 373–380.
- Ohguchi, H.; Harada, T.; Sagawa, M.; Kikuchi, S.; Tai, Y.T.; Richardson, P.G.; Hideshima, T.; Anderson, K.C. KDM6B modulates MAPK pathway mediating multiple myeloma cell growth and survival. Leukemia 2017, 31, 2661–2669.
- De Santa, F.; Totaro, M.G.; Prosperini, E.; Notarbartolo, S.; Testa, G.; Natoli, G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 2007, 130, 1083–1094.
- Varga-Weisz, P. ATP-dependent chromatin remodeling factors: Nucleosome shufflers with many missions. Oncogene 2001, 20, 3076–3085.
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422.
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492.
- Ogryzko, V.V.; Schiltz, R.L.; Russanova, V.; Howard, B.H.; Nakatani, Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 1996, 87, 953–959.
- Chen, H.P.; Zhao, Y.T.; Zhao, T.C. Histone deacetylases and mechanisms of regulation of gene expression. Crit. Rev. Oncog. 2015, 20, 35–47.
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840.
- Deng, S.; Hu, Q.; Zhang, H.; Yang, F.; Peng, C.; Huang, C. HDAC3 Inhibition Upregulates PD-L1 Expression in B-Cell Lymphomas and Augments the Efficacy of Anti-PD-L1 Therapy. Mol. Cancer Ther. 2019, 18, 900–908.
- Maharaj, K.; Powers, J.J.; Mediavilla-Varela, M.; Achille, A.; Gamal, W.; Quayle, S.; Jones, S.S.; Sahakian, E.; Pinilla-Ibarz, J. HDAC6 Inhibition Alleviates CLL-Induced T-Cell Dysfunction and Enhances Immune Checkpoint Blockade Efficacy in the Emu-TCL1 Model. Front. Immunol. 2020, 11, 590072.
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357.
- Stark, G.R.; Wang, Y.; Lu, T. Lysine methylation of promoter-bound transcription factors and relevance to cancer. Cell Res. 2011, 21, 375–380.
- Shi, Y.J.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear arnine oxidase homolog LSD1. Cell 2004, 119, 941–953.
- Kaniskan, H.U.; Jin, J. Recent progress in developing selective inhibitors of protein methyltransferases. Curr. Opin. Chem. Biol. 2017, 39, 100–108.
- Schafer, V.; Ernst, J.; Rinke, J.; Winkelmann, N.; Beck, J.F.; Hochhaus, A.; Gruhn, B.; Ernst, T. EZH2 mutations and promoter hypermethylation in childhood acute lymphoblastic leukemia. J. Cancer Res. Clin. Oncol. 2016, 142, 1641–1650.
- Santos-Rosa, H.; Schneider, R.; Bannister, A.J.; Sherriff, J.; Bernstein, B.E.; Emre, N.C.T.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Active genes are tri-methylated at K4 of histone H3. Nature 2002, 419, 407–411.
- Lachner, M.; O’Sullivan, R.J.; Jenuwein, T. An epigenetic road map for histone lysine methylation. J. Cell Sci. 2003, 116, 2117–2124.
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.H.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326.
- Bernhart, S.H.; Kretzmer, H.; Holdt, L.M.; Juhling, F.; Ammerpohl, O.; Bergmann, A.K.; Northoff, B.H.; Doose, G.; Siebert, R.; Stadler, P.F.; et al. Changes of bivalent chromatin coincide with increased expression of developmental genes in cancer. Sci. Rep. 2016, 6, 37393.
- Yin, S.; Yang, J.; Lin, B.; Deng, W.; Zhang, Y.; Yi, X.; Shi, Y.; Tao, Y.; Cai, J.; Wu, C.I.; et al. Exome sequencing identifies frequent mutation of MLL2 in non-small cell lung carcinoma from Chinese patients. Sci. Rep. 2014, 4, 6036.
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349.
- Hansen, K.H.; Bracken, A.P.; Pasini, D.; Dietrich, N.; Gehani, S.S.; Monrad, A.; Rappsilber, J.; Lerdrup, M.; Helin, K. A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol. 2008, 10, 1291–1300.
- Su, I.H.; Basavaraj, A.; Krutchinsky, A.N.; Hobert, O.; Ullrich, A.; Chait, B.T.; Tarakhovsky, A. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat. Immunol. 2003, 4, 124–131.
- Velichutina, I.; Shaknovich, R.; Geng, H.; Johnson, N.A.; Gascoyne, R.D.; Melnick, A.M.; Elemento, O. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood 2010, 116, 5247–5255.
- Hong, S.; Cho, Y.W.; Yu, L.R.; Yu, H.; Veenstra, T.D.; Ge, K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc. Natl. Acad. Sci. USA 2007, 104, 18439–18444.
- Agger, K.; Cloos, P.A.C.; Rudkjaer, L.; Williams, K.; Andersen, G.; Christensen, J.; Helin, K. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Gene Dev. 2009, 23, 1171–1176.
- Anderton, J.A.; Bose, S.; Vockerodt, M.; Vrzalikova, K.; Wei, W.; Kuo, M.; Helin, K.; Christensen, J.; Rowe, M.; Murray, P.G.; et al. The H3K27me3 demethylase, KDM6B, is induced by Epstein-Barr virus and over-expressed in Hodgkin’s Lymphoma. Oncogene 2011, 30, 2037–2043.
- Mallaney, C.; Ostrander, E.L.; Celik, H.; Kramer, A.C.; Martens, A.; Kothari, A.; Koh, W.K.; Haussler, E.; Iwamori, N.; Gontarz, P.; et al. Kdm6b regulates context-dependent hematopoietic stem cell self-renewal and leukemogenesis. Leukemia 2019, 33, 2506–2521.
- Li, Y.N.; Zhang, M.Y.; Sheng, M.Y.; Zhang, P.; Chen, Z.Z.; Xing, W.; Bai, J.; Cheng, T.; Yang, F.C.; Zhou, Y. Therapeutic potential of GSK-J4, a histone demethylase KDM6B/JMJD3 inhibitor, for acute myeloid leukemia. J. Cancer Res. Clin. 2018, 144, 1065–1077.
- Wang, W.; Lim, K.G.; Feng, M.; Bao, Y.; Lee, P.L.; Cai, Y.; Chen, Y.; Zhang, H.; Marzese, D.; Hoon, D.S.B.; et al. KDM6B Counteracts EZH2-Mediated Suppression of IGFBP5 to Confer Resistance to PI3K/AKT Inhibitor Treatment in Breast Cancer. Mol. Cancer Ther. 2018, 17, 1973–1983.
- Lagunas-Rangel, F.A. KDM6B (JMJD3) and its dual role in cancer. Biochimie 2021, 184, 63–71.
- Zhao, X.; Lwin, T.; Silva, A.; Shah, B.; Tao, J.; Fang, B.; Zhang, L.; Fu, K.; Bi, C.; Li, J.; et al. Unification of de novo and acquired ibrutinib resistance in mantle cell lymphoma. Nat. Commun. 2017, 8, 14920.
- Okkenhaug, K.; Burger, J.A. Erratum to: PI3K Signaling in Normal B Cells and Chronic Lymphocytic Leukemia (CLL). Curr. Top. Microbiol. Immunol. 2016, 393, E1.
- Vanhaesebroeck, B.; Leevers, S.J.; Ahmadi, K.; Timms, J.; Katso, R.; Driscoll, P.C.; Woscholski, R.; Parker, P.J.; Waterfield, M.D. Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem. 2001, 70, 535–602.
- Rudelius, M.; Pittaluga, S.; Nishizuka, S.; Pham, T.H.; Fend, F.; Jaffe, E.S.; Quintanilla-Martinez, L.; Raffeld, M. Constitutive activation of Akt contributes to the pathogenesis and survival of mantle cell lymphoma. Blood 2006, 108, 1668–1676.
- Herishanu, Y.; Katz, B.Z.; Lipsky, A.; Wiestner, A. Biology of chronic lymphocytic leukemia in different microenvironments: Clinical and therapeutic implications. Hematol. Oncol. Clin. N. Am. 2013, 27, 173–206.
- Spangle, J.M.; Roberts, T.M.; Zhao, J.J. The emerging role of PI3K/AKT-mediated epigenetic regulation in cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 123–131.
- Ortiz-Maldonado, V.; Garcia-Morillo, M.; Delgado, J. The biology behind PI3K inhibition in chronic lymphocytic leukaemia. Ther. Adv. Hematol. 2015, 6, 25–36.
- Daitoku, H.; Sakamaki, J.; Fukamizu, A. Regulation of FoxO transcription factors by acetylation and protein-protein interactions. Biochim. Biophys. Acta 2011, 1813, 1954–1960.
- Niimi, K.; Kohara, M.; Sedoh, E.; Fukumoto, M.; Shibata, S.; Sawano, T.; Tashiro, F.; Miyazaki, S.; Kubota, Y.; Miyazaki, J.I.; et al. FOXO1 regulates developmental lymphangiogenesis by upregulating CXCR4 in the mouse-tail dermis. Development 2020, 147.
- Zhou, X.R.; Li, X.; Liao, L.P.; Han, J.; Huang, J.; Li, J.C.; Tao, H.R.; Fan, S.J.; Chen, Z.F.; Li, Q.; et al. P300/CBP inhibition sensitizes mantle cell lymphoma to PI3Kdelta inhibitor idelalisib. Acta Pharmacol. Sin. 2021.
- Ravichandran, M.; Jurkowska, R.Z.; Jurkowski, T.P. Target specificity of mammalian DNA methylation and demethylation machinery. Org. Biomol. Chem. 2018, 16, 1419–1435.
- Sun, L.; Zhao, H.; Xu, Z.; Liu, Q.; Liang, Y.; Wang, L.; Cai, X.; Zhang, L.; Hu, L.; Wang, G.; et al. Phosphatidylinositol 3-kinase/protein kinase B pathway stabilizes DNA methyltransferase I protein and maintains DNA methylation. Cell Signal 2007, 19, 2255–2263.
- Zhao, H.F.; Zhang, L.; Guo, S.Q.; Yuan, T.; Xia, B.; Zhang, L.Y.; Zhang, Y.Z. Overexpression of DNA methyltransferase 1 as a negative independent prognostic factor in primary gastrointestinal diffuse large B-cell lymphoma treated with CHOP-like regimen and rituximab. Oncol. Lett. 2015, 9, 2307–2312.
- Trowbridge, J.J.; Snow, J.W.; Kim, J.; Orkin, S.H. DNA methyltransferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell 2009, 5, 442–449.
- Cha, T.L.; Zhou, B.P.; Xia, W.; Wu, Y.; Yang, C.C.; Chen, C.T.; Ping, B.; Otte, A.P.; Hung, M.C. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science 2005, 310, 306–310.
- Chartomatsidou, E.; Ntoufa, S.; Kotta, K.; Rovida, A.; Akritidou, M.A.; Belloni, D.; Ferrero, E.; Trangas, T.; Stavroyianni, N.; Anagnostopoulos, A.; et al. Inhibition of EZH2 and immune signaling exerts synergistic antitumor effects in chronic lymphocytic leukemia. Blood Adv. 2019, 3, 1891–1896.
- Szurian, K.; Csala, I.; Marosvari, D.; Rajnai, H.; Derso, K.; Bodor, C.; Piurko, V.; Matolcsy, A.; Reiniger, L. EZH2 is upregulated in the proliferation centers of CLL/SLL lymph nodes. Exp. Mol. Pathol. 2018, 105, 161–165.
- Pastore, A.; Gaiti, F.; Lu, S.X.; Brand, R.M.; Kulm, S.; Chaligne, R.; Gu, H.C.; Huang, K.Y.; Stamenova, E.K.; Beguelin, W.; et al. Corrupted coordination of epigenetic modifications leads to diverging chromatin states and transcriptional heterogeneity in CLL. Nat. Commun. 2019, 10.
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034.
- Neo, W.H.; Lim, J.F.; Grumont, R.; Gerondakis, S.; Su, I.H. c-Rel regulates Ezh2 expression in activated lymphocytes and malignant lymphoid cells. J. Biol. Chem. 2014, 289, 31693–31707.
- Kahl, B.S.; Spurgeon, S.E.; Furman, R.R.; Flinn, I.W.; Coutre, S.E.; Brown, J.R.; Benson, D.M.; Byrd, J.C.; Peterman, S.; Cho, Y.; et al. A phase 1 study of the PI3Kdelta inhibitor idelalisib in patients with relapsed/refractory mantle cell lymphoma (MCL). Blood 2014, 123, 3398–3405.
- Okkenhaug, K.; Burger, J.A. PI3K Signaling in Normal B Cells and Chronic Lymphocytic Leukemia (CLL). Curr. Top. Microbiol. Immunol. 2016, 393, 123–142.
- De Rooij, M.F.; Kuil, A.; Kater, A.P.; Kersten, M.J.; Pals, S.T.; Spaargaren, M. Ibrutinib and idelalisib synergistically target BCR-controlled adhesion in MCL and CLL: A rationale for combination therapy. Blood 2015, 125, 2306–2309.
- Mondello, P.; Derenzini, E.; Asgari, Z.; Philip, J.; Brea, E.J.; Seshan, V.; Hendrickson, R.C.; de Stanchina, E.; Scheinberg, D.A.; Younes, A. Dual inhibition of histone deacetylases and phosphoinositide 3-kinase enhances therapeutic activity against B cell lymphoma. Oncotarget 2017, 8, 14017–14028.


