 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kevin Verhoeff | + 1487 word(s) | 1487 | 2021-06-22 03:33:55 | | | |
| 2 | Nora Tang | Meta information modification | 1487 | 2021-06-25 02:45:49 | | | | |
| 3 | Conner Chen | Meta information modification | 1487 | 2021-09-22 04:14:50 | | |
Video Upload Options
Islets of Langerhan are a crucial group of cells that enable the metabolization, physiologic control, and utilization of glucose, the primary energy source for cells. In situ physiologic intraportal hormone delivery from the pancreatic islets of Langerhans maintains basal normoglycemia with insulin and counterbalances hypoglycemia with glucagon. Insulin output can increase up ten-fold after a meal, and return rapidly to basal levels with no hysteresis. Type 1 diabetes represents an increasing and growingly financially unsustainable disease occurring due to the destruction of pancreatic islets of Langerhans. Current injectable insulin technologies fail to recreate physiologic glycemic control that is managed by islet cells resulting in a tight 1–2 mmol/L glycemic variance. In our opinion, exogenous subcutaneous insulin delivery, even when provided by the most ideal closed loop systems, cannot recreate this degree of dynamic control. Current therapies fail to adequately achieve euglycemia, leading to significant diabetes complications and a risk of mortality. Thus, developing a cell-based cure for type 1 diabetes through islet cell generation and transplantation remains an ideal to strive for. Achieving this goal, especially with stem cell therapies, as demonstrated by the Edmonton protocol (Shapiro 2000), demands complete understanding of embryological differentiation and physiology of the islets of Langerhans.
1. Embryological Development and Structure
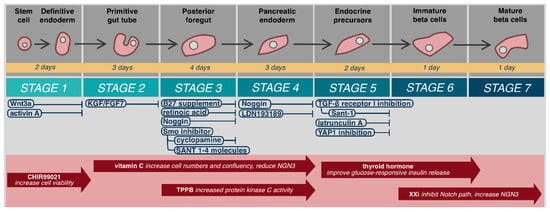
2. Function
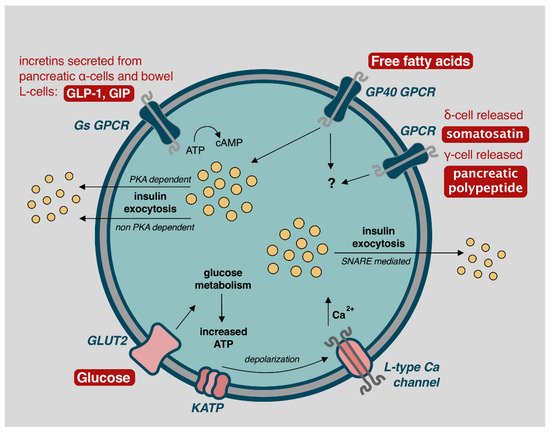
References
- Ionescu-Tirgoviste, C.; Gagniuc, P.A.; Gubceac, E.; Mardare, L.; Popescu, I.; Dima, S.; Militaru, M. A 3D map of the islet routes throughout the healthy human pancreas. Sci. Rep. 2015, 5, 14634.
- Rezania, A.; Bruin, J.E.; Riedel, M.J.; Mojibian, M.; Asadi, A.; Xu, J.; Gauvin, R.; Narayan, K.; Karanu, F.; O’Neil, J.J.; et al. Maturation of human embryonic stem cell-derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes 2012, 61, 2016–2029.
- Hogrebe, N.J.; Augsornworawat, P.; Maxwell, K.G.; Velazco-Cruz, L.; Millman, J.R. Targeting the cytoskeleton to direct pancreatic differentiation of human pluripotent stem cells. Nature Biotechnol. 2020, 38, 460–470.
- Rezania, A.; Bruin, J.E.; Arora, P.; Rubin, A.; Batushansky, I.; Asadi, A.; O’Dwyer, S.; Quiskamp, N.; Mojibian, M.; Albrecht, T.; et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 1121–1133.
- Shook, D.; Keller, R. Mechanisms, mechanics and function of epithelial-mesenchymal transitions in early development. Mech. Dev. 2003, 120, 1351–1383.
- Wang, H.; Ren, Y.; Hu, X.; Ma, M.; Wang, X.; Liang, H.; Liu, D. Effect of Wnt Signaling on the Differentiation of Islet β-Cells from Adipose-Derived Stem Cells. Biomed. Res. Int. 2017, 2017, 2501578.
- Kroon, E.; Martinson, L.A.; Kadoya, K.; Bang, A.G.; Kelly, O.G.; Eliazer, S.; Young, H.; Richardson, M.; Smart, N.G.; Cunningham, J.; et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 2008, 26, 443–452.
- Vincent, S.D.; Dunn, N.R.; Hayashi, S.; Norris, D.P.; Robertson, E.J. Cell fate decisions within the mouse organizer are governed by graded Nodal signals. Genes Dev. 2003, 17, 1646–1662.
- Brennan, J.; Lu, C.C.; Norris, D.P.; Rodriguez, T.A.; Beddington, R.S.; Robertson, E.J. Nodal signalling in the epiblast patterns the early mouse embryo. Nature 2001, 411, 965–969.
- Lowe, L.A.; Yamada, S.; Kuehn, M.R. Genetic dissection of nodal function in patterning the mouse embryo. Development 2001, 128, 1831.
- D’Amour, K.A.; Agulnick, A.D.; Eliazer, S.; Kelly, O.G.; Kroon, E.; Baetge, E.E. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nature Biotechnol. 2005, 23, 1534–1541.
- de Caestecker, M. The transforming growth factor-beta superfamily of receptors. Cytokine Growth Factor Rev. 2004, 15, 1–11.
- Kubo, A.; Shinozaki, K.; Shannon, J.M.; Kouskoff, V.; Kennedy, M.; Woo, S.; Fehling, H.J.; Keller, G. Development of definitive endoderm from embryonic stem cells in culture. Development 2004, 131, 1651–1662.
- Sui, L.; Leibel, R.L.; Egli, D. Pancreatic Beta Cell Differentiation From Human Pluripotent Stem Cells. Curr. Protoc. Hum. Genet. 2018, 99, e68.
- Chen, J.K.; Taipale, J.; Young, K.E.; Maiti, T.; Beachy, P.A. Small molecule modulation of Smoothened activity. Proc. Natl. Acad. Sci. USA 2002, 99, 14071.
- Mfopou, J.K.; Chen, B.; Mateizel, I.; Sermon, K.; Bouwens, L. Noggin, retinoids, and fibroblast growth factor regulate hepatic or pancreatic fate of human embryonic stem cells. Gastroenterology 2010, 138, 2233–2245.
- Hart, A.; Papadopoulou, S.; Edlund, H. Fgf10 maintains notch activation, stimulates proliferation, and blocks differentiation of pancreatic epithelial cells. Dev. Dyn. 2003, 228, 185–193.
- Pagliuca, F.W.; Millman, J.R.; Gurtler, M.; Segel, M.; Van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of functional human pancreatic beta cells in vitro. Cell 2014, 159, 428–439.
- Johansson, K.A.; Dursun, U.; Jordan, N.; Gu, G.; Beermann, F.; Gradwohl, G.; Grapin-Botton, A. Temporal control of neurogenin3 activity in pancreas progenitors reveals competence windows for the generation of different endocrine cell types. Dev. Cell 2007, 12, 457–465.
- Mamidi, A.; Prawiro, C.; Seymour, P.A.; de Lichtenberg, K.H.; Jackson, A.; Serup, P.; Semb, H. Mechanosignalling via integrins directs fate decisions of pancreatic progenitors. Nature 2018, 564, 114–118.
- Yabe, S.G.; Fukuda, S.; Takeda, F.; Nashiro, K.; Shimoda, M.; Okochi, H. Efficient generation of functional pancreatic β-cells from human induced pluripotent stem cells. J. Diabetes 2017, 9, 168–179.
- Rukstalis, J.M.; Habener, J.F. Neurogenin3: A master regulator of pancreatic islet differentiation and regeneration. Islets 2009, 1, 177–184.
- Suzuki, T.; Dai, P.; Hatakeyama, T.; Harada, Y.; Tanaka, H.; Yoshimura, N.; Takamatsu, T. TGF-β Signaling Regulates Pancreatic β-Cell Proliferation through Control of Cell Cycle Regulator p27 Expression. Acta Histochem. Cytochem. 2013, 46, 51–58.
- Chen, S.; Borowiak, M.; Fox, J.L.; Maehr, R.; Osafune, K.; Davidow, L.; Lam, K.; Peng, L.F.; Schreiber, S.L.; Rubin, L.L.; et al. A small molecule that directs differentiation of human ESCs into the pancreatic lineage. Nat. Chem. Biol. 2009, 5, 258–265.
- Rezania, A.; Bruin, J.E.; Xu, J.; Narayan, K.; Fox, J.K.; O’Neil, J.J.; Kieffer, T.J. Enrichment of human embryonic stem cell-derived NKX6.1-expressing pancreatic progenitor cells accelerates the maturation of insulin-secreting cells in vivo. STEM CELLS 2013, 31, 2432–2442.
- Aguayo-Mazzucato, C.; Zavacki, A.M.; Marinelarena, A.; Hollister-Lock, J.; El Khattabi, I.; Marsili, A.; Weir, G.C.; Sharma, A.; Larsen, P.R.; Bonner-Weir, S. Thyroid hormone promotes postnatal rat pancreatic β-cell development and glucose-responsive insulin secretion through MAFA. Diabetes 2013, 62, 1569–1580.
- Komatsu, M.; Takei, M.; Ishii, H.; Sato, Y. Glucose-stimulated insulin secretion: A newer perspective. J. Diabetes Investig. 2013, 4, 511–516.
- Thorens, B. Neural regulation of pancreatic islet cell mass and function. Diabetes Obes. Metab. 2014, 16, 87–95.
- Gilon, P.; Henquin, J.C. Mechanisms and physiological significance of the cholinergic control of pancreatic beta-cell function. Endocr. Rev. 2001, 22, 565–604.
- Latres, E.; Finan, D.A.; Greenstein, J.L.; Kowalski, A.; Kieffer, T.J. Navigating Two Roads to Glucose Normalization in Diabetes: Automated Insulin Delivery Devices and Cell Therapy. Cell Metab. 2019, 29, 545–563.
- Seino, S.; Shibasaki, T. PKA-Dependent and PKA-Independent Pathways for cAMP-Regulated Exocytosis. Physiol. Rev. 2005, 85, 1303–1342.
- Itoh, Y.; Kawamata, Y.; Harada, M.; Kobayashi, M.; Fujii, R.; Fukusumi, S.; Ogi, K.; Hosoya, M.; Tanaka, Y.; Uejima, H.; et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 2003, 422, 173–176.
- Capozzi, M.E.; Svendsen, B.; Encisco, S.E.; Lewandowski, S.L.; Martin, M.D.; Lin, H.; Jaffe, J.L.; Coch, R.W.; Haldeman, J.M.; MacDonald, P.E.; et al. beta Cell tone is defined by proglucagon peptides through cAMP signaling. JCI Insight 2019, 4.
- Li, N.; Yang, Z.; Li, Q.; Yu, Z.; Chen, X.; Li, J.-C.; Li, B.; Ning, S.-L.; Cui, M.; Sun, J.-P.; et al. Ablation of somatostatin cells leads to impaired pancreatic islet function and neonatal death in rodents. Cell Death Dis. 2018, 9, 682.
- Aslam, M.; Vijayasarathy, K.; Talukdar, R.; Sasikala, M.; Nageshwar Reddy, D. Reduced pancreatic polypeptide response is associated with early alteration of glycemic control in chronic pancreatitis. Diabetes Res. Clin. Pract. 2020, 160, 107993.
- Rabiee, A.; Galiatsatos, P.; Salas-Carrillo, R.; Thompson, M.J.; Andersen, D.K.; Elahi, D. Pancreatic polypeptide administration enhances insulin sensitivity and reduces the insulin requirement of patients on insulin pump therapy. J. Diabetes Sci. Technol. 2011, 5, 1521–1528.


