1. Embryological Development and Structure
Islets form collections of cells that exist uniformly throughout the pancreas but represent only 1–4%, 2 g, or 2 mL of the pancreatic volume [
28]. Person-to-person heterogeneity is common but islets are generally composed of approximately 60% β-cells, 30% α-cells, <10% δ-cells, <5% γ and ε cells producing insulin, glucagon, somatostatin, pancreatic polypeptide, and grehlin, respectively [
28]. Islet mass varies throughout life, and expands during childhood growth and during normal pregnancy. The exact mechanisms that regulate this expansion process remain incompletely understood.
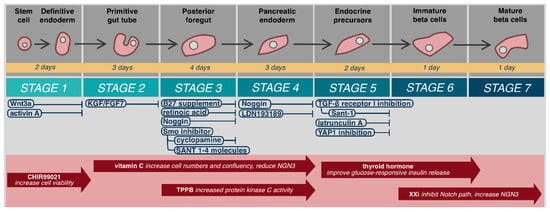
Mature β-cells develop from embryonic stem cells (hESC) in a continual process that may be considered in seven steps beginning from definitive endoderm, to primitive gut tube, posterior foregut, pancreatic endoderm, endocrine precursors, immature β-cells, and finally mature β-cells () [
29,
30,
31]. Definitive endoderm forms during gastrulation from epiblast cells undergoing epithelial to mesenchymal transition [
32]. This process is initiated by Wnt3a protein signaling [
29,
31,
33,
34], followed by Nodal signaling-mediated activation of the TGFβ pathway that ultimately leads to activation of intracellular Smad2 and differentiation into the primitive streak and definitive endoderm [
35,
36,
37]. Stable, bioactive Nodal does not exist. Fortunately, a similar protein from the TGFβ family, activin-A, acts as an in vitro biochemical analogue to activate Smad2 [
38,
39,
40]. In vitro, hESC exposure to Wnt3a and activin-A leads to 95% definitive endoderm cells that express the phenotypic markers SOX17 and FOXA2 [
41]. Patterning of anterior–posterior axis occurs with exposure to KGF/FGF7 and creates the primitive gut tube [
29,
30,
34]. Subsequent culture with B27 supplement, retinoic acid, Noggin, and a smoothened (Smo) inhibitor, such as cyclopamine or Sant 1–4 molecules to prevent Hedgehog (Hh) signaling, induces differentiation into the posterior foregut that has potential to become pancreatic, hepatic, or duodenal tissues [
29,
34,
42]. Hepatic tissues are favored through bone morphogenetic protein (BMP) signaling pathways, while endocrine differentiation is blocked by FGF10 activation [
43,
44]. Exposure to Noggin or LDN193189, potent inhibitors of both BMP and FGF10, produces pancreatic endoderm cells (PDX1
+) [
29,
34,
43].
Figure 1. Embryological differentiation and maturation of islet cells.
Further differentiation of pancreatic endoderm cells into islets has been incompletely understood and until recently, only occurred in three-dimensional (3D) culture in vitro [
31,
41,
45]. Differentiation into pancreatic endocrine progenitors (PDX1
+/NKX6.1
+) utilizes TGFβ receptor I (TβRI/ALK5) inhibition and continued prevention of Hh signaling with Sant1–4 moecules [
30,
31,
41]. Recent data has helped clarify why 3D culture and in vivo differentiation is required at this stage for β-cell differentiation. Failure to produce NKX6.1
+ cells prior to expression of endocrine genes such as neurogenin 3 (NEUROG3) produces non-functional poly-hormonal cells [
30,
46]. Hogrebe et al. (2020) recently demonstrated that the cellular microenvironment, actin cytoskeleton, and cellular attachments, dictate NEUROG3 expression [
30]. Firm adhesion of stage 4 (PDX1
+) cells to Type-I collagen coated culture plates leads to NKX6.1
+ cells, followed by stage 5 actin depolymerization with latrunculin A to allow NEUROG3 expression. Similarly, inhibition of YAP1 function increases NEUROG3 expression and favors endocrinogenesis [
47]. Further maturation leads to insulin producing, NKX6.1 expressing, β-cells with islet-like glucose response in vivo [
30].
Maturation and differentiation specificity and efficiency may be further improved with various compounds (). It should be noted that use and timing of these compounds varies widely by protocol. CHIR99021, a selective glycogen synthase kinase-3β inhibitor, has been used in stage 1 formation of definitive endoderm to increase cell viability [
30,
31,
48]. Rezania et al. added vitamin C from the primitive gut tube to pancreatic endoderm (stages 2–4), to increase cell numbers and confluency and reduce NGN3 expression, which has demonstrated disruption of pancreatic endoderm [
31,
49]. Increased protein kinase C activity, in vitro with -(
2S,5S)-(
E,E)-8-(5-(4-(trifluoromethyl)phenyl)-2,4-pentadienoylamino)benzolactam (TPPB) has demonstrated improved induction of pancreatic progenitors from primitive gut tube (stages 3–4) [
29,
30,
50,
51,
52]. Thyroid hormone acts after stage 5 through the transcription factor MAFA to improve glucose-responsive insulin release in mature cells [
30,
31,
41,
53]. Alongside thyroid hormone, gamma secretase XX inhibitor (XXi), which inhibits the Notch pathway and increases NGN3 expression, has been used in step 6 to inhibit PTF1a guided exocrine differentiation to improve β-cell maturation [
30,
31,
38,
41,
43,
49]. Application of these compounds is not standardized, and no author to date has combined all these additives to determine if an ideal, more efficient or specific β-cell differentiation can be achieved. Greater understanding of their role, result replication, and process standardization are needed to determine ideal additive compounds.
A major limitation in our understanding of islet developmental science is that many of the concepts and protocols have been derived from work in murine models—mainly because the relevant human targets and growth factors have yet to be defined. While there may be conservation in the early developmental pathways between species, it seems unlikely that this process will be fully optimized until these pathways are mapped out entirely in human cells. Another important limitation of in vitro islet generation is that it only approximates but does not replicate the continuum of cell-to-cell contact, dynamic intracellular signaling and participation of the physiologic extracellular matrix present in the full complexity of a developing human embryo. Only when we can recapitulate the process with more accuracy will we be able to optimize, perfect and avoid risk of off-target cell growth in this differentiation process.
2. Function
Glucose control is accomplished with both autonomic nervous and hormonal systems (). While interest focuses on β-cells, the α, δ, γ, and ε cells also play increasingly well understood and important roles in glycemic control.
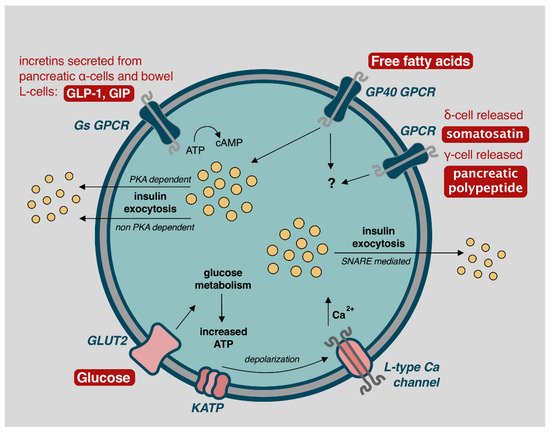
Figure 2. Mechanisms of β-cell insulin release and glycemic control. Image adapted from Komatsu et al. (2013) with permission for reuse [
56].
In the fasting state, normoglycemia is achieved through activation of the autonomic nervous system; sympathetic activation leads to glucagon release from α-cells, while parasympathetic activity induces insulin release from β-cells [
54]. These actions are directed through glucose-sensing cells located in peripheral locations such as the hepatoportal vein area, and by specialized glucose-excited or glucose-inhibited neurons located in the hypothalamus or brainstem region [
54]. This mechanism directs α and β-cells to release basal levels of glucagon and insulin to promote appropriate hepatic gluconeogenesis for anabolism and cellular functions [
54]. In anticipation of food, either by sight, mastication, or gastric distention, and prior to any blood glucose changes, parasympathetic release of acetylcholine activates β-cell muscarinic receptors (m3AchR), producing phospholipid-derived messengers to initiate protein kinase C (PKC) directed calcium influx and efficient insulin release through the cephalic response [
54,
55].
Elevated blood glucose concentrations lead to biphasic insulin release, lasting approximately 60 min [
5,
56]. The first phase occurs with GLUT2 facilitated diffusion of glucose into β-cells, which is oxidatively metabolized to produce ATP. In response, ATP dependent K
+ channels (KATP) channels close, leading to cellular membrane depolarization and opening of voltage-dependent L-type calcium channels. Intracellular calcium promotes SNARE protein mediated exocytosis of insulin-containing secretory granules with release into portal circulation () [
5,
56]. Depolarization and exocytosis oscillate every 3–6 min to avoid insulin receptor downregulation [
5,
56].
A second phase of insulin release, accounting for approximately 50% of postprandial insulin secretion, occurs via stimulation from parasympathetic inputs, glucagon-like peptide 1 (GLP-1), glucose-dependent insulinotropic peptide (GIP), free fatty acids (FFA), and somatostatin () [
5,
56,
57,
58,
59]. GLP-1 and GIP are incretins secreted from pancreatic α-cells, as well as K-cells and L-cells located in the pancreas, ileum, and colonic bowel in response to increase blood glucose concentration [
56,
59]. This demonstrates the α-cell interaction with β-cells to achieve euglycemia. GLP-1 and GIP act through β-cell G-protein-coupled receptors (GPCR), increasing 3′,5′-cyclic adenosine monophosphate (cAMP) and leading to protein kinase A (PKA) dependent and non-PKA dependent insulin exocytosis [
57,
59]. Similarly, FFA act through the GP40 GPCR to further stimulate insulin release [
57,
58]. δ-cell released somatostatin, and γ-cell released pancreatic polypeptide, also play a minor role for glucose homeostasis but mechanisms for such are incompletely understood [
60,
61]. Ablation of δ-cells impairs islet cell function [
60], and infusion of pancreatic polypeptide alongside insulin reduces insulin requirements [
62]—further analysis of these mechanisms may assist with improving glycemic control but also highlight the complex interplay of cells required for glycemic control that is often overlooked with single or dual hormone treatment systems.