 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Grazyna Kochan | + 2039 word(s) | 2039 | 2021-05-25 08:19:26 | | | |
| 2 | Nora Tang | Meta information modification | 2039 | 2021-06-24 09:38:49 | | | | |
| 3 | Nora Tang | Meta information modification | 2039 | 2021-06-25 10:13:19 | | |
Video Upload Options
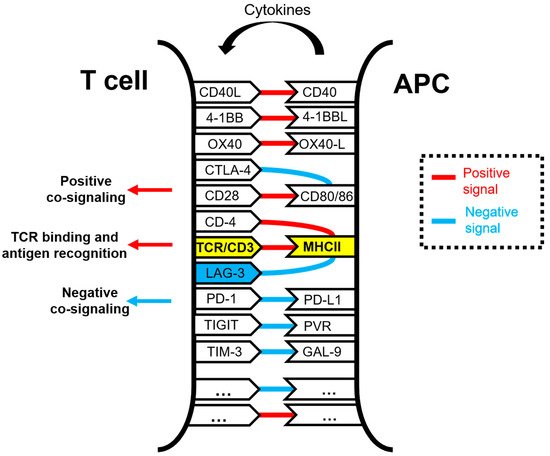
Lymphocyte activation gene 3 (LAG-3) is a cell surface inhibitory receptor with multiple biological activities over T cell activation and effector functions. LAG-3 plays a regulatory role in immunity and emerged some time ago as an inhibitory immune checkpoint molecule comparable to PD-1 and CTLA-4 and a potential target for enhancing anti-cancer immune responses. LAG-3 is the third inhibitory receptor to be exploited in human anti-cancer immunotherapies, and it is considered a potential next-generation cancer immunotherapy target in human therapy, right next to PD-1 and CTLA-4. Unlike PD-1 and CTLA-4, the exact mechanisms of action of LAG-3 and its relationship with other immune checkpoint molecules remain poorly understood. This is partly caused by the presence of non-conventional signaling motifs in its intracellular domain that are different from other conventional immunoregulatory signaling motifs but with similar inhibitory activities.
1. Function
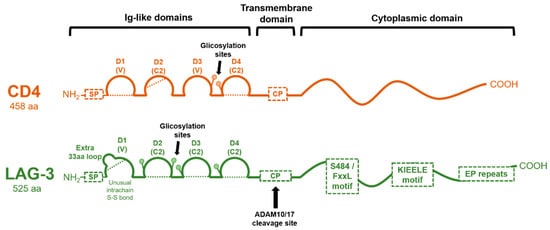
2. Genetic Structure of the LAG-3 Gene and Domain Organization
3. LAG-3 Ligands
References
- Huard, B.; Tournier, M.; Hercend, T.; Triebel, F.; Faure, F. Lymphocyte-Activation Gene 3/Major Histocompatibility Complex Class II Interaction Modulates the Antigenic Response of CD4+ T Lymphocytes. Eur. J. Immunol. 1994, 24, 3216–3221.
- Huard, B.; Prigent, P.; Pagès, F.; Bruniquel, D.; Triebel, F. T Cell Major Histocompatibility Complex Class II Molecules Down-Regulate CD4+ T Cell Clone Responses Following LAG-3 Binding. Eur. J. Immunol. 1996, 26, 1180–1186.
- Workman, C.J.; Dugger, K.J.; Vignali, D.A.A. Cutting Edge: Molecular Analysis of the Negative Regulatory Function of Lymphocyte Activation Gene-3. J. Immunol. 2002, 169, 5392–5395.
- Workman, C.J.; Rice, D.S.; Dugger, K.J.; Kurschner, C.; Vignali, D.A.A. Phenotypic Analysis of the Murine CD4-Related Glycoprotein, CD223 (LAG-3). Eur. J. Immunol. 2002, 32, 2255–2263.
- Workman, C.J.; Cauley, L.S.; Kim, I.-J.; Blackman, M.A.; Woodland, D.L.; Vignali, D.A.A. Lymphocyte Activation Gene-3 (CD223) Regulates the Size of the Expanding T Cell Population Following Antigen Activation in vivo. J. Immunol. 2004, 172, 5450–5455.
- Maçon-Lemaître, L.; Triebel, F. The Negative Regulatory Function of the Lymphocyte-Activation Gene-3 Co-Receptor (CD223) on Human T Cells. Immunology 2005, 115, 170–178.
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A.A. LAG3 (CD223) as a Cancer Immunotherapy Target. Immunol. Rev. 2017, 276, 80–96.
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a Novel Lymphocyte Activation Gene Closely Related to CD4. J. Exp. Med. 1990, 171, 1393–1405.
- Baixeras, B.E.; Huard, B.; Miossec, C.; Jitsukawa, S.; Martin, M.; Hercend, T.; Auffray, C.; Triebel, F.; Tonneau-Piatier, D. Characterization of the Lymphocyte Activation Gene 3-Encoded Protein. A New Ligand for Human Leukoc3~ Antigen Class H Antigens. Pharm. Res. 1992, 176, 327–337.
- Annunziato, F.; Manetti, R.; Tomasévic, I.; Giudizi, M.; Biagiotti, R.; Giannò, V.; Germano, P.; Mavilia, C.; Maggi, E.; Romagnani, S. Expression and Release of LAG-3-Encoded Protein by Human CD4 + T Cells Are Associated with IFN-Γ Production. FASEB J. 1996, 10, 769–776.
- Avice, M.N.; Sarfati, M.; Triebel, F.; Delespesse, G.; Demeure, C.E. Lymphocyte Activation Gene-3, a MHC Class II Ligand Expressed on Activated T Cells, Stimulates TNF-Alpha and IL-12 Production by Monocytes and Dendritic Cells. J. Immunol. 1999, 162, 2748–2753.
- Slevin, S.M.; Garner, L.C.; Lahiff, C.; Tan, M.; Wang, L.M.; Ferry, H.; Greenaway, B.; Lynch, K.; Geremia, A.; Hughes, S.; et al. Lymphocyte Activation Gene (LAG)-3 Is Associated with Mucosal Inflammation and Disease Activity in Ulcerative Colitis. J. Crohn’s Colitis. 2020, 14, 1446–1461.
- Hannier, S.; Tournier, M.; Bismuth, G.; Triebel, F. CD3/TCR Complex-Associated Lymphocyte Activation Gene-3 Molecules Inhibit CD3/TCR Signaling. J. Immunol. 1998, 161, 4058–4065.
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.A.; Wherry, E.J. Coregulation of CD8+ T Cell Exhaustion by Multiple Inhibitory Receptors During Chronic Viral Infection. Nat. Immunol. 2009, 10, 29–37.
- Chihara, N.; Madi, A.; Kondo, T.; Zhang, H.; Acharya, N.; Singer, M.; Nyman, J.; Marjanovic, N.D.; Kowalczyk, M.S.; Wang, C.; et al. Induction and Transcriptional Regulation of the Co-Inhibitory Gene Module in T Cells. Nature 2018, 558, 454–459.
- Grosso, J.F.; Kelleher, C.C.; Harris, T.J.; Maris, C.H.; Hipkiss, E.L.; De Marzo, A.; Anders, R.; Netto, G.; Derese, G.; Bruno, T.C.; et al. LAG-3 Regulates CD8 + T Cell Accumulation and Effector Function in Murine Self—and Tumor-Tolerance Systems. J. Clin. Invest. 2007, 117, 3383–3392.
- Grosso, J.F.; Goldberg, M.V.; Getnet, D.; Bruno, T.C.; Yen, H.-R.; Pyle, K.J.; Hipkiss, E.; Vignali, D.A.A.; Pardoll, D.M.; Drake, C.G. Functionally Distinct LAG-3 and PD-1 Subsets on Activated and Chronically Stimulated CD8 T Cells. J. Immunol. 2009, 182, 6659–6669.
- Huang, R.-Y.; Eppolito, C.; Lele, S.; Shrikant, P.; Matsuzaki, J.; Odunsi, K. LAG3 And PD1 Co-Inhibitory Molecules Collaborate to Limit CD8+ T Cell Signaling and Dampen Antitumor Immunity in a Murine Ovarian Cancer Model. Oncotarget 2015, 6, 27359.
- Williams, J.B.; Horton, B.L.; Zheng, Y.; Duan, Y.; Powell, J.D.; Gajewski, T.F. The EGR2 Targets LAG-3 and 4-1BB Describe and Regulate Dysfunctional Antigen-Specific CD8 + T Cells in the Tumor Microenvironment. J. Exp. Med. 2017, 214, 381–400.
- Huard, B.; Mastrangeli, R.; Prigent, P.; Bruniquel, D.; Donini, S.; El-Tayar, N.; Maigret, B.; Dreano, M.; Triebel, F. Characterization of the Major Histocompatibility Complex Class II Binding Site on LAG-3 Protein. Proc. Natl. Acad. Sci. USA 1997, 94, 5744–5749.
- Li, N.; Workman, C.J.; Martin, S.M.; Vignali, D.A.A. Biochemical Analysis of the Regulatory T Cell Protein Lymphocyte Activation Gene-3 (LAG-3; CD223). J. Immunol. 2004, 173, 6806–6812.
- Li, N.; Wang, Y.; Forbes, K.; Vignali, K.M.; Heale, B.S.; Saftig, P.; Hartmann, D.; Black, R.A.; Rossi, J.J.; Blobel, C.P.; et al. Metalloproteases Regulate T-Cell Proliferation and Effector Function Via LAG-3. EMBO J. 2007, 26, 494–504.
- Mastrangeli, R.; Micangeli, E.; Donini, S. Cloning of Murine LAG-3 by Magnetic Bead Bound Homologous Probes and PCR (GENE-CAPTURE PCR). Anal. Biochem. 1996, 241, 93–102.
- Workman, C.J.; Vignali, D.A.A. The CD4-Related Molecule, LAG-3 (CD223), Regulates the Expansion of Activated T Cells. Eur. J. Immunol. 2003, 3, 970–979.
- Maeda, T.K.; Sugiura, D.; Okazaki, M.T.; Okazaki, T. Atypical Motifs in the Cytoplasmic Region of the Inhibitory Immune Co-Receptor LAG-3 Inhibit T Cell Activation. J. Biol. Chem. 2019, 294, 6017–6026.
- Iouzalen, N.; Andreae, S.; Hannier, S.; Triebel, F. LAP, A Lymphocyte Activation Gene-3 (LAG-3)-Associated Protein that Binds to a Repeated EP Motif in the Intracellular Region of LAG-3, May Participate in the Down-Regulation of the CD3/TCR Activation Pathway. Eur. J. Immunol. 2001, 31, 2885–2891.
- Bae, J.; Lee, S.J.; Park, C.-G.; Lee, Y.S.; Chun, T. Trafficking of LAG-3 to the Surface on Activated T Cells via Its Cytoplasmic Domain and Protein Kinase C Signaling. J. Immunol. 2014, 193, 3101–3112.
- Long, L.; Zhang, X.; Chen, F.; Pan, Q.; Phiphatwatchara, P. The Promising Immune Checkpoint LAG-3: From Tumor Microenvironment to Cancer Immunotherapy. Genes Cancer 2018, 9, 176.
- Huard, B.; Prigent, P.; Tournier, M.; Bruniquel, D.; Triebel, F. CD4/Major Histocompatibility Complex Class II Interaction Analyzed with CD4—and Lymphocyte Activation Gene-3 (LAG-3)-Ig Fusion Proteins. Eur. J. Immunol. 1995, 25, 2718–2721.
- Hemon, P.; Jean-Louis, F.; Ramgolam, K.; Brignone, C.; Viguier, M.; Bachelez, H.; Triebel, F.; Charron, D.; Aoudjit, F.; Al-Daccak, R.; et al. MHC Class II Engagement by Its Ligand LAG-3 (CD223) Contributes to Melanoma Resistance to Apoptosis. J. Immunol. 2011, 186, 5173–5183.
- Donia, M.; Andersen, R.; Kjeldsen, J.W.; Fagone, P.; Munir, S.; Nicoletti, F.; Andersen, M.H.; Straten, P.T.; Svane, I.M. Aberrant Expression of MHC Class II in Melanoma Attracts Inflammatory Tumor-Specific CD4+T-Cells, which Dampen CD8+T-Cell Antitumor Reactivity. Cancer Res. 2015, 75, 3747–3759.
- Chung, L.Y.; Tang, S.J.; Wu, Y.C.; Sun, G.H.; Liu, H.Y.; Sun, K.H. Galectin-3 Augments Tumor Initiating Property and Tumorigenicity pf Lung Cancer through Interaction with Β-Catenin. Oncotarget 2015, 6, 4936–4952.
- Lu, W.; Wang, J.; Yang, G.; Yu, N.; Huang, Z.; Xu, H.; Li, J.; Qiu, J.; Zeng, X.; Chen, S.; et al. Posttranscriptional Regulation of Galectin-3 by miR—128 Contributes to Colorectal Cancer Progression. Oncotarget 2017, 8, 15242–15251.
- Li, M.; Feng, Y.M.; Fang, S.Q. Overexpression of Ezrin and Galectin-3 as Predictors of Poor Prognosis of Cervical Cancer. Braz. J. Med. Biol. Res. 2017, 50.
- Kouo, T.; Huang, L.; Pucsek, A.B.; Cao, M.; Solt, S.; Armstrong, T.; Jaffee, E. Galectin-3 Shapes Antitumor Immune Responses by Suppressing CD8+ T Cells via LAG-3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunol. Res. 2015, 3, 412–423.
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, W.; et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 2019, 176, 334–347.e12.

