1. Function
LAG-3 (CD223) is an inhibitory receptor first described in
in vitro-activated T cells. LAG-3 has multiple biological effects on the function of T cells and CD4 T cell activation, usually inhibitory [
1,
2]. LAG-3 does not seem to inhibit T cell function in the absence of CD4 activation but may interfere with CD4 coreceptor function. LAG-3 negatively regulates proliferation, activation, effector function, and homeostasis of both CD8 and CD4 T cells, as shown in LAG-3 knockout mice and by LAG-3 blockade with antibodies in human cells [
1,
3,
4,
5]. Thus, the evidence suggests that LAG-3 interferes with a common pathway to both CD4 and CD8 activation and regulates the activation and expansion of T memory cells [
6].
LAG-3 plays a regulatory role in the immune system comparable to PD-1 and CTLA-4, generally consisting of inhibition of cell proliferation, immune function, cytokine secretion, and homeostasis [
1,
2,
3,
7]. LAG-3 can be upregulated under various antigen stimulation conditions [
3,
8,
9]. Its expression is induced by TCR stimulation or cytokine stimulation, and LAG-3 is upregulated within activated, cytokine-expressing T cells [
9,
10,
11,
12]. LAG-3 associates with the TCR:CD3 complex at the T cell membrane and negatively regulates TCR signal transduction, which in turn terminates cell proliferation and cytokine secretion in response to CD3 signaling [
13]. LAG-3 and CD3 co-engagement in the immunological synapse is necessary to down-modulate TCR signal transduction. Among other consequences, the simultaneous engagement of LAG-3/TCR with their ligands inhibits TCR:CD3-dependent intracellular calcium fluxes. Similarly to PD-1, constitutive LAG-3 expression is frequently associated with exhausted T cells, and it is generally regarded as an exhaustion marker for CD4 and CD8 T cells in response to repetitive antigen stimulation in cancer and chronic viral infections [
14,
15,
16,
17,
18,
19]. Indeed, LAG-3 has been found to physically associate with TCR in both CD8 and CD4 T cells following TCR engagement, downregulating TCR-dependent signaling cascades and thereby dampening T cell responses [
13] ().
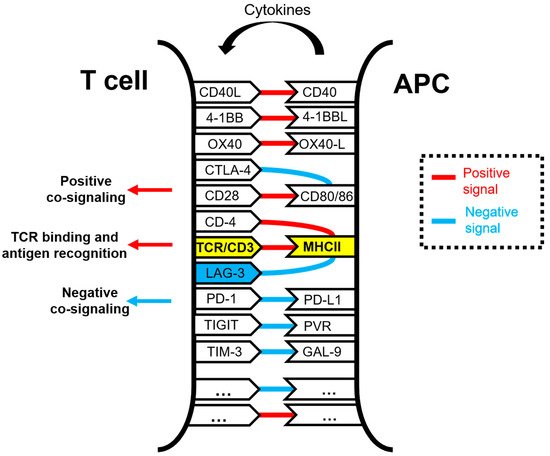
Figure 1. Schematic representation of the molecular interactions occurring within the immunological synapse between a T cell and an antigen-presenting cell (APC) during antigen presentation and T cell activation. The TCR/CD3 and MHC complexes are highlighted in yellow. Some of the well-known co-stimulatory and co-inhibitory receptor–ligand interactions are shown in the figure, linked by red lines for activating interactions and with blue lines for inhibitory interactions. Antigenic peptides presented by APCs loaded in MHC molecules are specifically recognized by the TCR, while other interactions take place simultaneously to deliver co-stimulatory and co-inhibitory signals. These interactions are integrated within T cells. The LAG-3 molecule is highlighted in blue.
2. Genetic Structure of the LAG-3 Gene and Domain Organization
Back in 1990, Dr. Frédéric Triebel and colleagues identified
lag-3 as a novel lymphocyte activation gene closely related to CD4. The authors found a 2-kb mRNA to be selectively transcribed in activated human T and NK cells [
8].
Lag-3 encodes a protein with a molecular weight of 70 kDa and resides on the distal part of the short arm of chromosome 12 (12p13.32) in humans and in chromosome 6 in mouse. The
lag-3 gene encodes a 498-amino acid type I membrane protein [
20]. Its locus is adjacent to that of the CD4 co-receptor and has a similar sequence and intron/exon organization, strongly indicating that
lag-3 and
CD4 genes have evolved by gene duplication from a pre-existing common evolutionary ancestor gene encoding a two-IgSF-domain structure.
The LAG-3 protein structure can be divided into extracellular, transmembrane, and intracellular regions (). The gene has 8 exons. Exon I encodes 19 aa of the hydrophobic leader peptide; exon II encodes 9 aa of the leader peptide (9 aa) and 41 aa of the extracellular region (41 aa); the rest of the extracellular region is encoded by exon III (101 aa), IV (90 aa), V (92 aa), and VI (81 aa); exon VII encodes the transmembrane region (44 aa); and exon VIII (21 aa), the highly charged cytoplasmic region [
8].
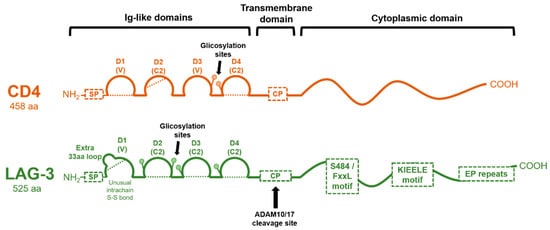
Figure 2. Molecular organization of CD4 and LAG-3 proteins. The domain organization of CD4 and LAG-3 is schematically shown in the figure, with each Ig-like domain indicated as arcs. Dotted lines represent disulfide bonds. The cleavage site for ADAM 10/17 in LAG-3 is shown, rendering a soluble version. CP: connecting peptide; SP: signal peptide.
The LAG-3 protein corresponds to a mature protein of 525 residues with a molecular mass of approximately 50 kDa. Both CD4 and LAG-3 extracellular regions are composed of four extracellular immunoglobulin superfamily-like (IgSF) domains (D1-D4) (). D1 is an Ig variable-like region (V-SET type), and it includes an extra, unique, proline-rich loop (30 residues) in the middle of the domain (between the C and C′ β strands of the D1 domain) and an unusual intra-chain di-sulfide bridge [
8]. Unlike CD4, this loop mediates LAG-3–MHCII interactions [
21]. Domains D2, D3, and D4 of LAG-3 belong to the C2-SET, while the CD4 D3 domain is of a V-SET type. D1 and D3 of LAG-3, as well as D2 and D4, share strong internal structural homologies. However, CD4 and LAG-3 only share less than 20% amino acid sequence homology. A β-strand between D1 and D2 and between D3 and D4 extends rigidly between these substructures [
20].
Additionally, compared to CD4, the LAG-3 protein has a longer connecting peptide between the membrane-proximal D4 domain and the transmembrane region. LAG-3 is cleaved within this connecting peptide, resulting in the release of a soluble form [
21]. This cleavage has been described to be mediated by two transmembrane metalloproteases, ADAM10 and ADAM17, regulated through two distinct mechanisms. While ADAM10 mediates constitutive LAG-3 cleavage, ADAM17 mediates LAG-3 cleavage induced by TCR signaling in a PKCθ-dependent manner [
22]. Interestingly, there is evidence that shedding of LAG-3 from the cell surface enhances T-cell proliferation and effector function, which adds another activating level of immune regulation by LAG-3 [
21,
22].
The cytoplasmic tail of LAG-3 mediates intracellular negative signal transduction, as its deletion completely abrogates its inhibitory function. The cytoplasmic tail contains three conserved motifs (): (1) a potentially phosphorylatable serine (S484), (2) a KIEELE motif, and (3) a glutamate-proline dipeptide multiple repeats motif (EP motif). Nevertheless, there is conflicting evidence on how these motifs affect LAG-3 molecular function and downstream signaling. The potential serine phosphorylation motif (S484) has been described within the FxxL motif, although no specific function has been ascribed to it other than its correlation with IL-2 production. This serine may be analogous to the protein kinase C binding site in the CD4 molecule [
23].
The KIEELE motif corresponds to a highly conserved short sequence not found in other proteins, and its relevance to LAG-3 function remains controversial. According to Workman et al., the deletion of KIEELE completely abrogates LAG-3 function on CD4 T cells, highlighting its importance for LAG-3 inhibitory signaling [
3,
24]. In addition, an alanine scanning mutagenesis of KIEELE residues showed that lysine residue (K468) was absolutely required for LAG-3 downstream signaling, with minor contributions by E470 and E471 in CD4 function. However, according to Maeda and colleagues, LAG-3 mediates a cell-intrinsic, negative inhibitory signal independently of KIEELE via two distinct mechanisms that are dependent on the FxxL motif in the membrane proximal region and the carboxy-terminal EP repeats [
25].
The EP repeat motif is present in some proteins in diverse species and cellular locations, which could be an indicator of a potentially common biological function. For example, PDGF-R, LckBP1, and SPY75 contain similar EP regions that contribute to their signaling mechanisms. It was proposed that the EP motif was key in counteracting the CD3/TCR activation pathway via association to LAP protein (LAG-3 associated protein), allowing LAG-3 co-localization with CD3, CD4, and CD8 within lipid rafts [
26]. According to Workman and colleagues, the deletion of the EP motif or the S484A mutation maintained LAG-3 activity and function in CD4
+ and CD4
− T cells, suggesting that these two features may not have been essential [
3]. Nevertheless, it has to be stressed that the inhibitory activity of LAG-3 was only completely abrogated in CD4
− T cells if both KIEELE and EP motifs were removed. The authors proposed that the EP motif could play a role in preventing LAG-3 from acting as a stimulatory co-receptor such as CD4 rather than cooperating with KIEELE in inhibitory signal transduction.
As LAG-3 is localized in lipid rafts following antigen stimulation [
26], its cytoplasmic domain could be mediating its location in the cell membrane. LAG-3 co-localizes with glycosphingolipid-enriched microdomain complexes containing CD3, CD4, or CD8. After TCR engagement, LAG-3 co-localizes with CD3 and ganglioside GM1 in the immunological synapse. However, no systematic follow-up studies on these motifs and their functions have been reported to date. The structural nature of LAG-3 needs to be elucidated to understand its mechanisms of action on the regulation of T cell activation.
Apart from its surface expression, LAG-3 can also be stored in lysosomes, and it is translocated to the membranes of activated T cells via its cytoplasmic domain by protein kinase C signaling [
27].
3. LAG-3 Ligands
MHC-II molecules are considered the canonical ligand of LAG-3, with which it stably interacts through the D1 domain with a significantly higher affinity than with CD4 [
20]. This association negatively regulates T cell activation, cytotoxicity, and cytokine production [
28]. In fact, LAG-3-Ig fusion proteins act as competitors in CD4/MHC class II-dependent cellular adhesion assays [
29]. Once LAG-3 is bound to MHC-II, it transmits inhibitory signals via its cytoplasmic domain and inhibits CD4 T cell activation [
1,
2]. Nevertheless, the molecular mechanisms of signal transduction remain broadly unknown compared to other immune checkpoint molecules. LAG-3:MHC class II binding contributes to tumor escape from apoptosis [
30] and facilitates recruitment of tumor-specific CD4 T cells, but with a reduction of the CD8 T cell response [
31].
Initially, the high-affinity LAG-3:MHC class II interaction was proposed to be the main mechanism of the inhibitory activities of LAG-3 through competition with CD4:MHC II binding [
1,
2]. However, it still remains controversial whether the interaction with MHC-II is solely responsible for mediating LAG-3 immunosuppressive functions, in light of the recent identification of additional ligands.
The second LAG-3 ligand to be described was galectin-3 (Gal-3), a 31 kDa galactose-binding lectin that modulates T cell activation. Gal-3 has been shown to be highly expressed in various tumor cells and in activated T lymphocytes [
32,
33,
34,
35]. Gal-3 binds to LAG-3 and appears to be required for optimal inhibition of CD8 T cell cytotoxic function. Gal-3 shapes antitumor-specific immune responses by suppressing activated antigen-committed CD8 T cells via LAG-3 expression in the tumor microenvironment and inhibiting expansion of plasmacytoid dendritic cells [
35].
The liver-secreted protein fibrinogen-like protein 1 (FGL1) was recently identified as a new LAG-3 functional ligand [
36]. FGL1 is a member of the fibrinogen family and possesses immunosuppressive activities by inhibiting antigen-specific T cells through LAG-3 binding. FGL1 interacts with LAG-3 via the fibrinogen-like domain with the D1 and D2 Ig-like domains of LAG-3. Its expression is induced by IL-6 and is highly up-regulated in several human cancers such us lung cancer, prostate cancer, melanoma, and colorectal cancer. Indeed, it was demonstrated that elevated plasma FGL1 levels were associated with poor cancer outcomes and anti-PD-1 therapy resistance [
36]. Thus, this binding was described as a novel mechanism of immune evasion, and its blockade potentiated anti-tumor T cell immunity in pre-clinical models. The FGL1/LAG-3 blockade may synergize with anti-PD-1/anti-PD-L1 therapy. A single point mutation (Y73F) in the D1 C’ strand domain of LAG-3 disrupted LAG-3/MHC-II binding [
3,
4,
20], but not FGL1-Ig binding. Nevertheless, this mutant LAG-3 could still inhibit T cells, demonstrating that FGL1/LAG-3 interactions correspond to a tumor immune evasion mechanism which is non-redundant to MHC-II binding.
Taken together, these data suggest that LAG-3 interactions with several ligands are important for its inhibitory function, and unlike what was thought before, LAG-3 does not function primarily by disrupting CD4:MHC-II interactions.