 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sébastien Deshayes | + 2536 word(s) | 2536 | 2021-05-24 10:30:59 | | | |
| 2 | Vivi Li | Meta information modification | 2536 | 2021-06-21 11:04:43 | | |
Video Upload Options
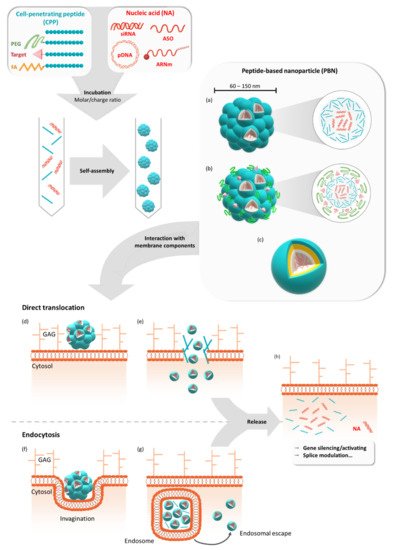
Gene therapy offers the possibility to skip, repair, or silence faulty genes or to stimulate the immune system to fight against disease by delivering therapeutic nucleic acids (NAs) to a patient. Compared to other drugs or protein treatments, NA-based therapies have the advantage of being a more universal approach to designing therapies because of the versatility of NA design. NAs (siRNA, pDNA, or mRNA) have great potential for therapeutic applications for an immense number of indications. However, the delivery of these exogenous NAs is still challenging and requires a specific delivery system. In this context, beside other non-viral vectors, cell-penetrating peptides (CPPs) gain more and more interest as delivery systems by forming a variety of nanocomplexes depending on the formulation conditions and the properties of the used CPPs/NAs.
1. Introduction
2. GALA/KALA/RALA Family
3. PepFect/NickFect Family
4. WRAP Family
Studies on CADY-K and RICK peptides have emphasized the requirement for several structural properties for both PBN formation and the resulting biological activity. As already observed for most amphipathic peptides, the existence of distinct hydrophobic and hydrophilic domains was required for cargo interactions, as well as for nanoparticle formation. In addition, the analysis of amino acid composition revealed a strong redundancy of arginine and tryptophan residues [57][58][59][60]. Based on this knowledge, a new family of CPPs was conceived: WRAP (W- and R- rich amphipathic peptides) were composed of only three amino acids (leucine, arginine, and tryptophan) [26]. These short (15/16mer) peptides were able to form stable PBNs, enroll siRNA in different cell lines (U87, MCF7, Neuro2a, HT29, etc.), and trigger more than 50% luciferase silencing at low siRNA concentrations (20–50 nM, depending on the cell line). This knock-down efficiency resulted from a rapid PBN internalization within 5–15 min of incubation.
Later on, the rapid internalization of the WRAP-PBNs was associated with their internalization mechanism [61]. By combining the whole panel of available approaches, including biophysical (leakage assay), biological (dynamin triple-KO cells), confocal (endocytosis and vesicle markers), and electron microscopy experiments, our laboratory could highlight that the balance between direct translocation and endocytosis-dependent internalization clearly shifted in favor of direct translocation through the plasma membrane. Furthermore, we deduced that the low percentage of endocytosis was mainly due to naturally occurring endocytosis processes at the surface of the cells. More interestingly, even if some percentage of WRAP-PBNs was internalized by endocytosis-dependent mechanisms, they could be able to rapidly escape from endosomes, as suggested by leakage assays using large unilamellar vesicles (LUVs) reflecting the endosomal membrane composition.
Recently, we performed a structure activity relationship (SAR) study using the lead peptides WRAP1 and WRAP5 and 13 new analogues to gain more information about the relationship between the amino acid composition, nanoparticle formation, and cellular internalization of these siRNA-loaded peptides (manuscript submitted for publication).
The WRAP5 peptide was also shown to be a suitable gene delivery system in the context of cancer gene therapy, as shown by the WRAP5-mediated delivery of a p53 encoding plasmid (pDNA) [40]. Through the design of an experimental tool, the optimal ratio of nitrogen to phosphate groups (N/P) was determined for WRAP5:pDNA in comparison with the complex formed by the previously presented RALA peptide. In this context, both peptides were able to form PBNs in the presence of pDNA, with nearly identical zeta potential (~ +33 mV) and pDNA complexation capacity (~90%), but with a smaller PBN size for WRAP5 compared to RALA (103.0 nm at N/P = 3 and 183.3 nm at N/P = 5, respectively).
5. Mgpe Family
Based on investigations modulating the amphipathicity and charges of several pVec analogues, Dr. M. Ganguli’s group modified the physicochemical parameters of the amphipathic peptide Mgpe-1, derived from human protein phosphatase 1E, to promote nucleic acid delivery [21][62]. The Mgpe family includes primary and secondary amphipathic peptides, mainly tested for plasmid delivery in different cell lines. Mgpe-3 and Mgpe-4 peptides displayed a high transfection efficiency, equivalent to that of commercial agents with a lower cytotoxicity and with stability in the presence of serum [21]. In addition, several developments have enabled the improvement of pDNA transfection efficacy. For example, the addition of cysteine increased the transfection efficiency of a secondary amphipathic Mgpe-9, and the coating of Mgpe/plasmid polyplexes with glycosaminoglycans such as chondroitin sulphate (CS) displayed the enhancement of polyplexes’ stability and pDNA delivery efficiency [23][63]. Recently, Ganguli and co-workers described that Mgpe polyplexes could also induce a high transgene expression in differentiated non-dividing cells, known to be difficult to transfect, and that an additional CS coating improved the diffusion of the polyplexes in the vitreous, suggesting the possibility of delivering genetic material to the retina [64].
References
- Hoy, S.M. Patisiran: First Global Approval. Drugs 2018, 78, 1625–1631.
- Scott, L.J. Givosiran: First Approval. Drugs 2020, 80, 335–339.
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 MRNA Covid-19 Vaccine. N. Engl. J. Med. 2020.
- Ledford, H. Moderna COVID Vaccine Becomes Second to Get US Authorization. Nature 2020.
- Crooke, S.T. Antisense Strategies. Curr. Mol. Med. 2004, 4, 465–487.
- Tolia, N.H.; Joshua-Tor, L. Slicer and the Argonautes. Nat. Chem. Biol. 2007, 3, 36–43.
- Whitehead, K.A.; Langer, R.; Anderson, D.G. Knocking down Barriers: Advances in SiRNA Delivery. Nat. Rev. Drug Discov. 2009, 8, 129–138.
- Reschke, C.R.; Henshall, D.C. MicroRNA and Epilepsy. Adv. Exp. Med. Biol. 2015, 888, 41–70.
- Gaspar, R.; Coelho, F.; Silva, B.F.B. Lipid-Nucleic Acid Complexes: Physicochemical Aspects and Prospects for Cancer Treatment. Molecules 2020, 25, 5006.
- Ginn, S.L.; Amaya, A.K.; Alexander, I.E.; Edelstein, M.; Abedi, M.R. Gene Therapy Clinical Trials Worldwide to 2017: An Update. J. Gene Med. 2018, 20, e3015.
- Pearson, S.; Jia, H.; Kandachi, K. China Approves First Gene Therapy. Nat. Biotechnol. 2004, 22, 3–4.
- Waldrop, M.A.; Kolb, S.J. Current Treatment Options in Neurology—SMA Therapeutics. Curr. Treat. Options Neurol. 2019, 21, 25.
- Langel, Ü. (Ed.) Cell-Penetrating Peptides; Methods in Molecular Biology; Springer New York: New York, NY, USA, 2015; Volume 1324, ISBN 978-1-4939-2805-7.
- Ramsey, J.D.; Flynn, N.H. Cell-Penetrating Peptides Transport Therapeutics into Cells. Pharmacol. Ther. 2015, 154, 78–86.
- Kauffman, W.B.; Fuselier, T.; He, J.; Wimley, W.C. Mechanism Matters: A Taxonomy of Cell Penetrating Peptides. Trends Biochem. Sci. 2015, 40, 749–764.
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in Oligonucleotide Drug Delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694.
- Boisguérin, P.; Deshayes, S.; Gait, M.J.; O’Donovan, L.; Godfrey, C.; Betts, C.A.; Wood, M.J.A.; Lebleu, B. Delivery of Therapeutic Oligonucleotides with Cell Penetrating Peptides. Adv. Drug Deliv. Rev. 2015, 87, 52–67.
- Lehto, T.; Ezzat, K.; Wood, M.J.A.; El Andaloussi, S. Peptides for Nucleic Acid Delivery. Adv. Drug Deliv. Rev. 2016, 106, 172–182.
- Kurrikoff, K.; Langel, Ü. Recent CPP-Based Applications in Medicine. Expert Opin. Drug Deliv. 2019, 16, 1183–1191.
- Morris, M.C.; Vidal, P.; Chaloin, L.; Heitz, F.; Divita, G. A New Peptide Vector for Efficient Delivery of Oligonucleotides into Mammalian Cells. Nucleic Acids Res. 1997, 25, 2730–2736.
- Sharma, R.; Shivpuri, S.; Anand, A.; Kulshreshtha, A.; Ganguli, M. Insight into the Role of Physicochemical Parameters in a Novel Series of Amphipathic Peptides for Efficient DNA Delivery. Mol. Pharm. 2013, 10, 2588–2600.
- Konate, K.; Crombez, L.; Deshayes, S.; Decaffmeyer, M.; Thomas, A.; Brasseur, R.; Aldrian, G.; Heitz, F.; Divita, G. Insight into the Cellular Uptake Mechanism of a Secondary Amphipathic Cell-Penetrating Peptide for SiRNA Delivery. Biochemistry 2010, 49, 3393–3402.
- Sharma, R.; Nisakar, D.; Shivpuri, S.; Ganguli, M. Contrasting Effects of Cysteine Modification on the Transfection Efficiency of Amphipathic Peptides. Biomaterials 2014, 35, 6563–6575.
- Andaloussi, S.E.L.; Lehto, T.; Mäger, I.; Rosenthal-Aizman, K.; Oprea, I.I.; Simonson, O.E.; Sork, H.; Ezzat, K.; Copolovici, D.M.; Kurrikoff, K.; et al. Design of a Peptide-Based Vector, PepFect6, for Efficient Delivery of SiRNA in Cell Culture and Systemically in Vivo. Nucleic Acids Res. 2011, 39, 3972–3987.
- Vaissière, A.; Aldrian, G.; Konate, K.; Lindberg, M.F.; Jourdan, C.; Telmar, A.; Seisel, Q.; Fernandez, F.; Viguier, V.; Genevois, C.; et al. A Retro-Inverso Cell-Penetrating Peptide for SiRNA Delivery. J. Nanobiotechnology 2017, 15, 34.
- Konate, K.; Dussot, M.; Aldrian, G.; Vaissière, A.; Viguier, V.; Neira, I.F.; Couillaud, F.; Vivès, E.; Boisguerin, P.; Deshayes, S. Peptide-Based Nanoparticles to Rapidly and Efficiently “Wrap ’n Roll” SiRNA into Cells. Bioconjug. Chem. 2019, 30, 592–603.
- Kurrikoff, K.; Gestin, M.; Langel, Ü. Recent in Vivo Advances in Cell-Penetrating Peptide-Assisted Drug Delivery. Expert Opin. Drug Deliv. 2016, 13, 373–387.
- Konate, K.; Lindberg, M.F.; Vaissiere, A.; Jourdan, C.; Aldrian, G.; Margeat, E.; Deshayes, S.; Boisguerin, P. Optimisation of Vectorisation Property: A Comparative Study for a Secondary Amphipathic Peptide. Int. J. Pharm. 2016, 509, 71–84.
- Li, W.; Nicol, F.; Szoka, F.C. GALA: A Designed Synthetic PH-Responsive Amphipathic Peptide with Applications in Drug and Gene Delivery. Adv. Drug Deliv. Rev. 2004, 56, 967–985.
- Subbarao, N.K.; Fielding, C.J.; Hamilton, R.L.; Szoka, F.C. Lecithin:Cholesterol Acyltransferase Activation by Synthetic Amphipathic Peptides. Proteins 1988, 3, 187–198.
- Plank, C.; Oberhauser, B.; Mechtler, K.; Koch, C.; Wagner, E. The Influence of Endosome-Disruptive Peptides on Gene Transfer Using Synthetic Virus-like Gene Transfer Systems. J. Biol. Chem. 1994, 269, 12918–12924.
- Wyman, T.B.; Nicol, F.; Zelphati, O.; Scaria, P.V.; Plank, C.; Szoka, F.C. Design, Synthesis, and Characterization of a Cationic Peptide That Binds to Nucleic Acids and Permeabilizes Bilayers. Biochemistry 1997, 36, 3008–3017.
- Lee, H.; Jeong, J.H.; Park, T.G. PEG Grafted Polylysine with Fusogenic Peptide for Gene Delivery: High Transfection Efficiency with Low Cytotoxicity. J. Controlled Release 2002, 79, 283–291.
- Lee, H.; Jeong, J.H.; Park, T.G. A New Gene Delivery Formulation of Polyethylenimine/DNA Complexes Coated with PEG Conjugated Fusogenic Peptide. J. Controlled Release 2001, 76, 183–192.
- Lim, D.W.; Yeom, Y.I.; Park, T.G. Poly(DMAEMA-NVP)-b-PEG-Galactose as Gene Delivery Vector for Hepatocytes. Bioconjug. Chem. 2000, 11, 688–695.
- Katayama, T.; Kinugawa, S.; Takada, S.; Furihata, T.; Fukushima, A.; Yokota, T.; Anzai, T.; Hibino, M.; Harashima, H.; Yamada, Y. A Mitochondrial Delivery System Using Liposome-Based Nanocarriers That Target Myoblast Cells. Mitochondrion 2019, 49, 66–72.
- McCarthy, H.O.; McCaffrey, J.; McCrudden, C.M.; Zholobenko, A.; Ali, A.A.; McBride, J.W.; Massey, A.S.; Pentlavalli, S.; Chen, K.-H.; Cole, G.; et al. Development and Characterization of Self-Assembling Nanoparticles Using a Bio-Inspired Amphipathic Peptide for Gene Delivery. J. Controlled Release 2014, 189, 141–149.
- McErlean, E.M.; McCrudden, C.M.; McBride, J.W.; Cole, G.; Kett, V.L.; Robson, T.; Dunne, N.J.; McCarthy, H.O. Rational Design and Characterisation of an Amphipathic Cell Penetrating Peptide for Non-Viral Gene Delivery. Int. J. Pharm. 2021, 596, 120223.
- McCrudden, C.M.; McBride, J.W.; McCaffrey, J.; McErlean, E.M.; Dunne, N.J.; Kett, V.L.; Coulter, J.A.; Robson, T.; McCarthy, H.O. Gene Therapy with RALA/INOS Composite Nanoparticles Significantly Enhances Survival in a Model of Metastatic Prostate Cancer. Cancer Nanotechnol. 2018, 9, 5.
- Sousa, Â.; Almeida, A.M.; Faria, R.; Konate, K.; Boisguerin, P.; Queiroz, J.A.; Costa, D. Optimization of Peptide-Plasmid DNA Vectors Formulation for Gene Delivery in Cancer Therapy Exploring Design of Experiments. Colloids Surf. B Biointerfaces 2019, 183, 110417.
- Bennett, R.; Yakkundi, A.; McKeen, H.D.; McClements, L.; McKeogh, T.J.; McCrudden, C.M.; Arthur, K.; Robson, T.; McCarthy, H.O. RALA-Mediated Delivery of FKBPL Nucleic Acid Therapeutics. Nanomedicine 2015, 10, 2989–3001.
- Mulholland, E.J.; Ali, A.; Robson, T.; Dunne, N.J.; McCarthy, H.O. Delivery of RALA/SiFKBPL Nanoparticles via Electrospun Bilayer Nanofibres: An Innovative Angiogenic Therapy for Wound Repair. J. Controlled Release 2019, 316, 53–65.
- Yan, L.-P.; Castaño, I.M.; Sridharan, R.; Kelly, D.; Lemoine, M.; Cavanagh, B.L.; Dunne, N.J.; McCarthy, H.O.; O’Brien, F.J. Collagen/GAG Scaffolds Activated by RALA-SiMMP-9 Complexes with Potential for Improved Diabetic Foot Ulcer Healing. Mater. Sci. Eng. C Mater. Biol. Appl. 2020, 114, 111022.
- Udhayakumar, V.K.; De Beuckelaer, A.; McCaffrey, J.; McCrudden, C.M.; Kirschman, J.L.; Vanover, D.; Van Hoecke, L.; Roose, K.; Deswarte, K.; De Geest, B.G.; et al. Arginine-Rich Peptide-Based MRNA Nanocomplexes Efficiently Instigate Cytotoxic T Cell Immunity Dependent on the Amphipathic Organization of the Peptide. Adv. Healthc. Mater. 2017, 6.
- Cole, G.; Ali, A.A.; McErlean, E.; Mulholland, E.J.; Short, A.; McCrudden, C.M.; McCaffrey, J.; Robson, T.; Kett, V.L.; Coulter, J.A.; et al. DNA Vaccination via RALA Nanoparticles in a Microneedle Delivery System Induces a Potent Immune Response against the Endogenous Prostate Cancer Stem Cell Antigen. Acta Biomater. 2019, 96, 480–490.
- Mäe, M.; El Andaloussi, S.; Lundin, P.; Oskolkov, N.; Johansson, H.J.; Guterstam, P.; Langel, U. A Stearylated CPP for Delivery of Splice Correcting Oligonucleotides Using a Non-Covalent Co-Incubation Strategy. J. Controlled Release 2009, 134, 221–227.
- Ezzat, K.; Andaloussi, S.E.L.; Zaghloul, E.M.; Lehto, T.; Lindberg, S.; Moreno, P.M.D.; Viola, J.R.; Magdy, T.; Abdo, R.; Guterstam, P.; et al. PepFect 14, a Novel Cell-Penetrating Peptide for Oligonucleotide Delivery in Solution and as Solid Formulation. Nucleic Acids Res. 2011, 39, 5284–5298.
- Ezzat, K.; Helmfors, H.; Tudoran, O.; Juks, C.; Lindberg, S.; Padari, K.; El-Andaloussi, S.; Pooga, M.; Langel, Ü. Scavenger Receptor-Mediated Uptake of Cell-Penetrating Peptide Nanocomplexes with Oligonucleotides. FASEB J. 2012, 26, 1172–1180.
- Lindberg, S.; Regberg, J.; Eriksson, J.; Helmfors, H.; Muñoz-Alarcón, A.; Srimanee, A.; Figueroa, R.A.; Hallberg, E.; Ezzat, K.; Langel, Ü. A Convergent Uptake Route for Peptide- and Polymer-Based Nucleotide Delivery Systems. J. Controlled Release 2015, 206, 58–66.
- Kurrikoff, K.; Veiman, K.-L.; Künnapuu, K.; Peets, E.M.; Lehto, T.; Pärnaste, L.; Arukuusk, P.; Langel, Ü. Effective in Vivo Gene Delivery with Reduced Toxicity, Achieved by Charge and Fatty Acid -Modified Cell Penetrating Peptide. Sci. Rep. 2017, 7, 17056.
- Gestin, M.; Helmfors, H.; Falato, L.; Lorenzon, N.; Michalakis, F.I.; Langel, Ü. Effect of Small Molecule Signaling in PepFect14 Transfection. PLoS ONE 2020, 15, e0228189.
- Van den Brand, D.; Gorris, M.A.J.; van Asbeck, A.H.; Palmen, E.; Ebisch, I.; Dolstra, H.; Hällbrink, M.; Massuger, L.F.A.G.; Brock, R. Peptide-Mediated Delivery of Therapeutic MRNA in Ovarian Cancer. Eur. J. Pharm. Biopharm. 2019, 141, 180–190.
- Freimann, K.; Arukuusk, P.; Kurrikoff, K.; Vasconcelos, L.D.F.; Veiman, K.-L.; Uusna, J.; Margus, H.; Garcia-Sosa, A.T.; Pooga, M.; Langel, Ü. Optimization of in Vivo DNA Delivery with NickFect Peptide Vectors. J. Controlled Release 2016, 241, 135–143.
- Arukuusk, P.; Pärnaste, L.; Oskolkov, N.; Copolovici, D.-M.; Margus, H.; Padari, K.; Möll, K.; Maslovskaja, J.; Tegova, R.; Kivi, G.; et al. New Generation of Efficient Peptide-Based Vectors, NickFects, for the Delivery of Nucleic Acids. Biochim. Biophys. Acta BBA - Biomembr. 2013, 1828, 1365–1373.
- Freimann, K.; Arukuusk, P.; Kurrikoff, K.; Pärnaste, L.; Raid, R.; Piirsoo, A.; Pooga, M.; Langel, Ü. Formulation of Stable and Homogeneous Cell-Penetrating Peptide NF55 Nanoparticles for Efficient Gene Delivery In Vivo. Mol. Ther. Nucleic Acids 2018, 10, 28–35.
- Kurrikoff, K.; Freimann, K.; Veiman, K.-L.; Peets, E.M.; Piirsoo, A.; Langel, Ü. Effective Lung-Targeted RNAi in Mice with Peptide-Based Delivery of Nucleic Acid. Sci. Rep. 2019, 9, 19926.
- Jafari, M.; Karunaratne, D.N.; Sweeting, C.M.; Chen, P. Modification of a Designed Amphipathic Cell-Penetrating Peptide and Its Effect on Solubility, Secondary Structure, and Uptake Efficiency. Biochemistry 2013, 52, 3428–3435.
- Bechara, C.; Pallerla, M.; Burlina, F.; Illien, F.; Cribier, S.; Sagan, S. Massive Glycosaminoglycan-Dependent Entry of Trp-Containing Cell-Penetrating Peptides Induced by Exogenous Sphingomyelinase or Cholesterol Depletion. Cell. Mol. Life Sci. CMLS 2015, 72, 809–820.
- Jobin, M.-L.; Blanchet, M.; Henry, S.; Chaignepain, S.; Manigand, C.; Castano, S.; Lecomte, S.; Burlina, F.; Sagan, S.; Alves, I.D. The Role of Tryptophans on the Cellular Uptake and Membrane Interaction of Arginine-Rich Cell Penetrating Peptides. Biochim. Biophys. Acta 2015, 1848, 593–602.
- Walrant, A.; Bauzá, A.; Girardet, C.; Alves, I.D.; Lecomte, S.; Illien, F.; Cardon, S.; Chaianantakul, N.; Pallerla, M.; Burlina, F.; et al. Ionpair-π Interactions Favor Cell Penetration of Arginine/Tryptophan-Rich Cell-Penetrating Peptides. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183098.
- Deshayes, S.; Konate, K.; Dussot, M.; Chavey, B.; Vaissière, A.; Van, T.N.N.; Aldrian, G.; Padari, K.; Pooga, M.; Vivès, E.; et al. Deciphering the Internalization Mechanism of WRAP:SiRNA Nanoparticles. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183252.
- Rajpal; Mann, A.; Khanduri, R.; Naik, R.J.; Ganguli, M. Structural Rearrangements and Chemical Modifications in Known Cell Penetrating Peptide Strongly Enhance DNA Delivery Efficiency. J. Controlled Release 2012, 157, 260–271.
- Nisakar, D.; Vij, M.; Pandey, T.; Natarajan, P.; Sharma, R.; Mishra, S.; Ganguli, M. Deciphering the Role of Chondroitin Sulfate in Increasing the Transfection Efficiency of Amphipathic Peptide-Based Nanocomplexes. ACS Biomater. Sci. Eng. 2019, 5, 45–55.
- Subia, B.; Reinisalo, M.; Dey, N.; Tavakoli, S.; Subrizi, A.; Ganguli, M.; Ruponen, M. Nucleic Acid Delivery to Differentiated Retinal Pigment Epithelial Cells Using Cell-Penetrating Peptide as a Carrier. Eur. J. Pharm. Biopharm. 2019, 140, 91–99.


