Gene therapy offers the possibility to skip, repair, or silence faulty genes or to stimulate the immune system to fight against disease by delivering therapeutic nucleic acids (NAs) to a patient. Compared to other drugs or protein treatments, NA-based therapies have the advantage of being a more universal approach to designing therapies because of the versatility of NA design. NAs (siRNA, pDNA, or mRNA) have great potential for therapeutic applications for an immense number of indications. However, the delivery of these exogenous NAs is still challenging and requires a specific delivery system. In this context, beside other non-viral vectors, cell-penetrating peptides (CPPs) gain more and more interest as delivery systems by forming a variety of nanocomplexes depending on the formulation conditions and the properties of the used CPPs/NAs.
1. Introduction
Since 2016, we have observed an acceleration in the development of nucleic acid as therapeutics with the approval of several molecules by the U.S. Food and Drug Administration (FDA). For example, two therapeutics based on RNA interference (RNAi) were approved, ONPATTRO
® (Partisiran) for polyneuropathy in hereditary transthyretin-mediated (hATTR) amyloidosis in 2018 [
1], and GIVLAARI™ (Givosiran) for acute hepatic porphyria (AHP) in 2019 [
2]. More recently, resulting from the worldwide COVID-19 pandemic, two vaccines based on mRNA technology were put on the market by the companies Pfizer/BioNTec [
3] and Moderna [
4].
Oligonucleotides (ONs) are short polymers of nucleic acids (RNAs or DNAs), which could be natural or chemically modified. The use of therapeutic ONs to treat a wide range of diseases has expanded the range of possible targets beyond what is generally accessible by conventional pharmaceutics, such as gene silencing, splice modulation, or gene activation. Several examples could be mentioned, such as the antisense oligonucleotides (ASOs, 15 to 20 nucleotides), acting primarily in the nucleus by selectively cleaving pre-mRNAs having complementary sites via an RNase H dependent mechanism [
5]. Subsequently, double-stranded short interfering RNAs (siRNAs) that contain 20–25 nucleotides were developed as major therapeutic tools for silencing gene expression. The double-stranded siRNA is separated by helicase, and the antisense strand (or guide strand) is embedded into the RNA-induced silencing complex (RISC) to guide it to the complementary target mRNA for degradation [
6,
7]. Micro RNAs (miRs) are an endogenous, highly conserved, small non-coding RNA composed of 20–24 nucleotides that have been implicated as key regulators of target gene expression [
8]. At the post-transcriptional level, miRs bind to the 3′-untranslated regions of the corresponding target mRNAs of protein-coding genes, thereby resulting in target mRNA degradation and the inhibition of mRNA translation.
Longer nucleic acids (NAs) such as therapeutic DNAs are mainly used in the form of plasmids (pDNA), which encode specific genes or regulatory sequences for endogenous proteins [
9]. In this context, the suppressor gene p53 is the most widely transferred gene in clinical trials due to the fact that it is one of the most frequently mutated genes in different types of cancer [
10]. In 2003, the Chinese company Shenzhen SiBiono GenTech obtained approval from the State Food and Drug Administration of China for its recombinant adenovirus-based p53 gene therapy for head and neck squamous cell carcinoma [
11]. Another example is the most recently authorized gene therapy drug by the U.S. Food and Drug Administration (May 2019) called Zolgensma (Novartis) for the expression of the survival motor neuron 1 protein (SMN1) in motor neurons for the treatment of spinal muscular atrophy (SMA) [
12]. pDNA can also be used to edit genes via the CRISPR mechanism through the internalization of plasmids coding the Cas9 protein and the RNA guide strand with the targeted gene sequence.
Despite significant advances in different therapeutic NA applications, a major obstacle preventing their widespread usage is the challenge of organ- and tissue-specific delivery. To overcome this bottleneck, several strategies have been employed such as the chemical modification of the nucleic acid to improve its ‘drug-likeness’, as well as the use of cell-targeting or cell-penetrating moieties for covalent conjugation or nanoparticle formulation. More than twenty years ago, cell-penetrating peptides (CPPs) were identified as potential carriers for a wide variety of biomolecules, including NAs [
13,
14,
15]. Usually defined as short (up to 30 amino acids) peptides that originate from different sources (e.g., humans, mice, viruses or purely synthetic), CPPs were developed as one of the most promising non-viral strategies for improving the intracellular routing of NAs, since they constitute a great alternative to the existing viral (adenoviruses, retrovirus, etc.), lipid-based, or polymer-based methods [
16].
Initially applied through covalent conjugation to NAs, CPPs were increasingly used in non-covalent strategies based on electrostatic and hydrophobic interactions between both CPPs and NAs (). These interactions resulted in the self-assembly of peptides with NAs and the formation of peptide-based nanoparticles (PBNs), thus opening peptides to the field of nanomedicine [
17,
18,
19]. The delivery of NAs by peptides has become a separated subfield in the research domain of CPPs due to the formation of larger intermolecular structures instead of the monomolecular solutions of covalent CPP-NA conjugates. Moreover, peptide-based vectors are now considered to be suitable candidates for the delivery of therapeutic NAs due to their easy automated synthesis, single-step formulation, and biocompatible properties.
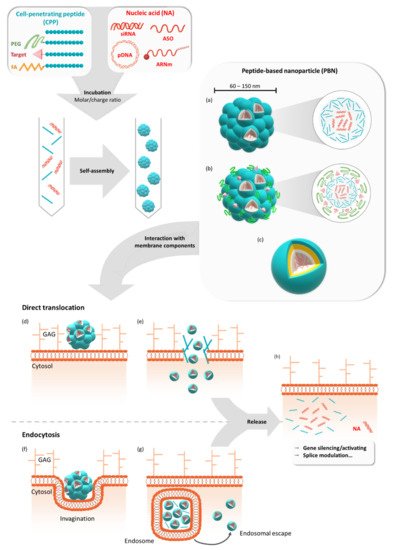
Figure 1. Formulation of peptide-based nanoparticles in the presence of different nucleic acids and their cellular internalization. Peptide-based nanoparticles (PBNs) are formulated by mixing a cell-penetrating peptide (CPP) or a grafted CPP (PEGylated, targeting sequence or fatty acid) with a nucleic acid (NA) such as pDNA, mRNA, siRNA, or ASO at a given molar or charge ratio. By mixing these two compounds, the nanoparticle is formed by self-assembling into naked PBNs (a), a multi-grafted PBNs (b), or prospective micelle-like PBNs (no model available) (c). In all cases, the PBNs of mean size between 60 nm and 150 nm encapsulate several NAs for cellular transfection. Thereafter, cellular internalization could occur via direct translocation (d) or via endocytosis-dependent pathways (f). After the direct translocation (e) or endosomal escape (g), the NAs could be active either by silencing or activating genes or by performing splice modulation (h). GAG = glycosaminoglycans.
Among the large number of CPPs, several amphipathic peptides were designed with both hydrophilic and hydrophobic domains in order to confer both NA complexation and membrane interaction abilities. Primary amphipathic CPPs have these two domains distributed according to each amino acid position along the peptide chain, as shown for Mgpe and MPG [
20,
21], while secondary amphipathic peptides result from the formation of both hydrophilic and hydrophobic domains through the secondary structure folding [
22,
23]. Many of the CPPs of this later class were used to form PBNs such as PepFect [
24], RICK [
25], or WRAP [
26]. However, although originally based on amphipathic peptides, the nanoparticles approach has been extended to all peptides and peptide analogues that were able to form stable CPP:NA nanoparticles and to improve NA delivery into mammalian cells [
19,
27]. The ability of CPPs to form PBNs has been associated with several structural properties. While the conformational state of non-covalent CPPs has been shown to play an important role in the interaction with NAs, as well as in the self-assembly process leading to efficient PBNs [
26,
28], the role of physicochemical parameters such as the amphipathicity, charges, and presence of specific residues is directly related to PBN efficiency [
21].
2. GALA/KALA/RALA Family
The 30 amino acid long, amphipathic, and α-helical GALA peptide [
74] was first designed as a lipid bilayer interactor at low pH due to its fusogenic properties [
75]. The bilayer destabilizing properties of GALA were used to promote gene delivery in vitro in combination with poly-lysine-conjugated ligands [
76]. To favor endosomal escape, GALA peptide was modified by replacing glutamate residues with lysine residues, resulting in the KALA peptide, which was also able to condense nucleic acids to nanoparticles [
77]. When environmental pH decreased from 7.5 to 5.0, KALA peptides undergo a pH-dependent amphipathic α-helix to random coil conformational change, leading to entrapped cargo release. KALA has also been used in combination with poly-lysine [
78], polyethylenimine (PEI) [
79], and (poly (DMAEMA-NVP))-b-PEG-galactose [
80] for DNA gene delivery. More recently, Katayama and co-workers found that a liposome modified with the KALA peptide was the most effective drug delivery system for mitochondrial targeting in C2C12 cells [
81].
Afterwards, the KALA peptide was further modified to improve transfection efficiency by changing the lysine residues to arginine residues [
34]. With seven arginine residues, the RALA peptide formed nanoparticles in the presence of anionic entities such as plasmids in a highly tunable way depending on the used molar peptide/DNA ratios, changing the size and surface charge of the PBNs. RALA PBNs were internalized via the clathrin- and caveolin-mediated endocytosis pathways, but a pH drop in the endosomes induced an increasing α-helicity of RALA, provoking the endosomal release of the transfected cargo. Modification of the RALA sequence in terms of amino acid composition and sequence length failed to improve the functional characteristics of RALA, confirming its superior sequence for non-toxic gene delivery [
82]. RALA is a widely used peptide-based delivery system, mainly optimized for the transfection of different oligonucleotides such as plasmids [
34,
83,
84], siRNA [
35,
36,
37], mRNA [
38], and for DNA vaccination [
85], demonstrating its broad utility.
3. PepFect/NickFect Family
Based on the short transportan-derived peptide TP10, Prof. Ü. Langel’s group developed a subset of different CPPs for NA delivery. In brief, TP10 was modified with stearic acid to improve non-covalent ON-complex formation and to enhance peptide-membrane interactions [
86]. This peptide, which was later named PepFect 3 (PF3), was used as a base for further modifications resulting in the widely studied analogues PepFect 6 (PF6) and PepFect 14 (PF14). The PF6 peptide was modified with endosomolytic trifluoromethylquinoline moiety, aiming to increase the endosomal escape of the peptide [
24]. The PF14 peptide was designed with non-encoded ornithine residues for increased stability and improved uptake [
39]. All PF complexes were described as being taken up via receptor-mediated endocytosis involving class-A scavenger receptors (SCARAs) [
87,
88].
More recently, based on physicochemical features in the complex formation and on the biological efficacy, a series of PF14 modifications were developed with altered charges and fatty acid contents. Kurrikoff and colleagues showed that with an optimal combination of overall charge and hydrophobicity in the peptide backbone, in vivo gene delivery can be enhanced [
42]. Interestingly, Gestin and co-workers found that through an optimized high-throughput luciferase assay, small molecule drugs (MPEP, VU0357121 and Ciproxifan) induced an increased transfection efficacy of PF14 complexed to splice-correcting oligonucleotides [
41]. This finding was quite surprising because it was not really clear whether the drugs influenced nanoparticle formation, and the underlying mechanism of cellular entry was not defined.
With regard to mRNA transfection, Prof. R. Brock’s group published the use of PepFect14 to formulate CPP-mRNA nanoparticles, showing efficient reporter protein expression in two- and three-dimensional cancer cell cultures [
40]. More importantly, following an intraperitoneal injection of PBNs encapsulating mCherry coding mRNA, they could reveal an important mCherry protein expression within the tumors of the treated mice. This protein expression was not observed in mice treated with the naked mRNA or with the mRNA transfected with Lipofectamine MessengerMax.
More or less in parallel to the PepFect family, Prof. Ü Langel’s group developed the NickFect family from the PF3 sequence [
44]. First, in order to enhance cellular uptake and endosomal release, the PF3 peptide was modified at Lys7, located within the linker between the galanin and the mastoparan residues (from the former Transportan or TP10 peptides) [
43]. In detail, by replacing Lys7 with ornithine and continuing the synthesis by coupling Gly6 to the δ-NH2 group of ornithine, the authors obtained the NickFect 51 (NF51) peptide-forming PBNs in the presence of pDNA, and were able to transfect different cell types. Based on this sequence, a novel amphipathic α-helical peptide, NF55, was designed for efficient in vivo DNA delivery by modifying the net charge and the helicity of the CPP [
44,
45]. More recently, Freimann and co-workers presented a new formulation approach called cryo-concentration for obtaining stable and homogeneous nanoparticles showing significantly higher bioactivity in vivo [
89].
4. WRAP Family
Studies on CADY-K and RICK peptides have emphasized the requirement for several structural properties for both PBN formation and the resulting biological activity. As already observed for most amphipathic peptides, the existence of distinct hydrophobic and hydrophilic domains was required for cargo interactions, as well as for nanoparticle formation. In addition, the analysis of amino acid composition revealed a strong redundancy of arginine and tryptophan residues [49,94–96]. Based on this knowledge, a new family of CPPs was conceived: WRAP (W- and R- rich amphipathic peptides) were composed of only three amino acids (leucine, arginine, and tryptophan) [26]. These short (15/16mer) peptides were able to form stable PBNs, enroll siRNA in different cell lines (U87, MCF7, Neuro2a, HT29, etc.), and trigger more than 50% luciferase silencing at low siRNA concentrations (20–50 nM, depending on the cell line). This knock-down efficiency resulted from a rapid PBN internalization within 5–15 min of incubation.
Later on, the rapid internalization of the WRAP-PBNs was associated with their internalization mechanism [97]. By combining the whole panel of available approaches, including biophysical (leakage assay), biological (dynamin triple-KO cells), confocal (endocytosis and vesicle markers), and electron microscopy experiments, our laboratory could highlight that the balance between direct translocation and endocytosis-dependent internalization clearly shifted in favor of direct translocation through the plasma membrane. Furthermore, we deduced that the low percentage of endocytosis was mainly due to naturally occurring endocytosis processes at the surface of the cells. More interestingly, even if some percentage of WRAP-PBNs was internalized by endocytosis-dependent mechanisms, they could be able to rapidly escape from endosomes, as suggested by leakage assays using large unilamellar vesicles (LUVs) reflecting the endosomal membrane composition.
Recently, we performed a structure activity relationship (SAR) study using the lead peptides WRAP1 and WRAP5 and 13 new analogues to gain more information about the relationship between the amino acid composition, nanoparticle formation, and cellular internalization of these siRNA-loaded peptides (manuscript submitted for publication).
The WRAP5 peptide was also shown to be a suitable gene delivery system in the context of cancer gene therapy, as shown by the WRAP5-mediated delivery of a p53 encoding plasmid (pDNA) [84]. Through the design of an experimental tool, the optimal ratio of nitrogen to phosphate groups (N/P) was determined for WRAP5:pDNA in comparison with the complex formed by the previously presented RALA peptide. In this context, both peptides were able to form PBNs in the presence of pDNA, with nearly identical zeta potential (~ +33 mV) and pDNA complexation capacity (~90%), but with a smaller PBN size for WRAP5 compared to RALA (103.0 nm at N/P = 3 and 183.3 nm at N/P = 5, respectively).
5. Mgpe Family
Based on investigations modulating the amphipathicity and charges of several pVec analogues, Dr. M. Ganguli’s group modified the physicochemical parameters of the amphipathic peptide Mgpe-1, derived from human protein phosphatase 1E, to promote nucleic acid delivery [21,100]. The Mgpe family includes primary and secondary amphipathic peptides, mainly tested for plasmid delivery in different cell lines. Mgpe-3 and Mgpe-4 peptides displayed a high transfection efficiency, equivalent to that of commercial agents with a lower cytotoxicity and with stability in the presence of serum [21]. In addition, several developments have enabled the improvement of pDNA transfection efficacy. For example, the addition of cysteine increased the transfection efficiency of a secondary amphipathic Mgpe-9, and the coating of Mgpe/plasmid polyplexes with glycosaminoglycans such as chondroitin sulphate (CS) displayed the enhancement of polyplexes’ stability and pDNA delivery efficiency [23,54]. Recently, Ganguli and co-workers described that Mgpe polyplexes could also induce a high transgene expression in differentiated non-dividing cells, known to be difficult to transfect, and that an additional CS coating improved the diffusion of the polyplexes in the vitreous, suggesting the possibility of delivering genetic material to the retina [55].
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines9050583