 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Christophe Furger | + 2653 word(s) | 2653 | 2021-06-15 05:34:49 | | | |
| 2 | Vivi Li | + 192 word(s) | 2845 | 2021-06-21 04:35:04 | | |
Video Upload Options
Plant extracts and pharmacopoeias represent an exceptional breeding ground for the discovery of new antioxidants. Until recently, the antioxidant activity was only measured by chemical hydrogen atom transfer (HAT) and single-electron transfer (SET) cell-free assays that do not inform about the actual effect of antioxidants in living systems. By providing information about the mode of action of antioxidants at the subcellular level, recently developed live cell assays are now changing the game.
1. Introduction
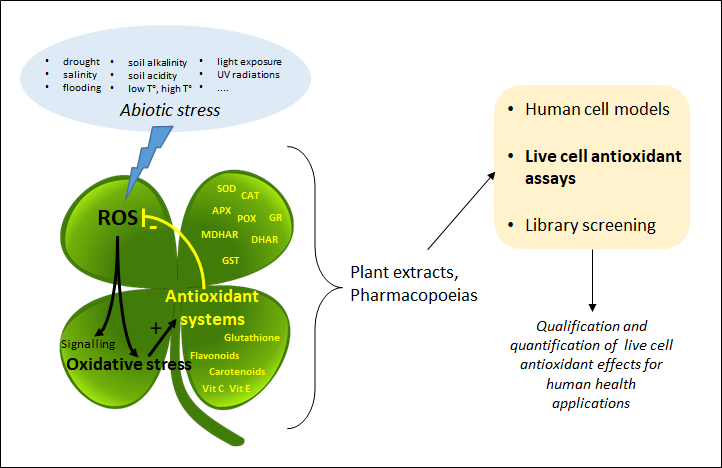
2. Cell Antioxidant Assay (CAA)
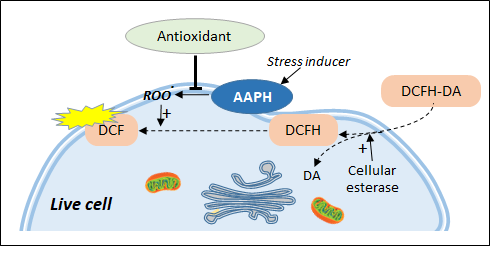
Figure 2. Live cell antioxidant assays based on chemical stress inducers. DCFH-DA is trapped in the cell in the form of DCFH which can be transformed by peroxidation products into the fluorescent DCF. Antioxidant effect is measured as the ability to inhibit the formation of AAPH-induced lipid peroxidation.
3. AOP1, a New Antioxidant Live Cell Approach Based on Photoinduction
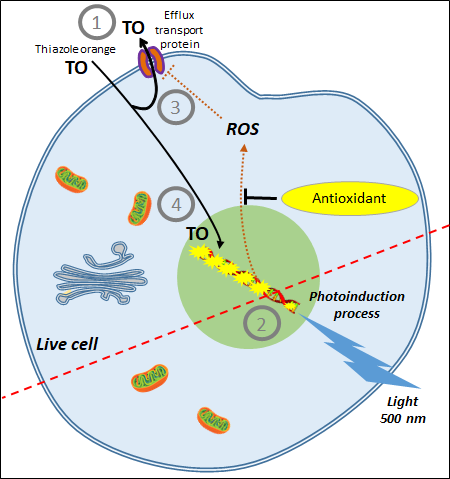
| CAA Assay | AOP1 Assay |
|---|---|
| Based on the production of AAPH-induced peroxyl radicals | Based on the controlled production of 1O2 and free radicals by photoinduction |
| Measures effects of plasma membrane-based antioxidants | Measures effects of intracellular-based antioxidants |
| No control of ROS production | Easy control of ROS production by light intensity; allows monitoring ROS production at a sublethal level (i.e., more physiological concentrations) |
| Interpretation limited to AAPH effects | |
| Does not differentiate between antioxidant and cytotoxic effects | Can easily discriminate between antioxidant and cytotoxic effects |
| Results need to be confirmed by performing a cytotoxicity assay (e.g., MTT) | No other assay needed |
| DCFH-DA subject to auto-oxidation | Sensor not directly involved in the oxidation process |
| Subject to cell leakage | No cell leakage |
| Fluorescence levels vary according to cell density | No effect of cell density (measure on a ratio mode) |
| Needs culture medium washes that disrupt cell culture | No washes required |
| Difficult to standardize | Easy to standardize |
| Detection by fluorescence readers | Detection by fluorescence readers + illuminator |
| Limited to adherent cells | Works for adherent and suspension cells, and organotypic models |
References
- Soares, C.; Carvalho, M.E.A.; Azevedo, R.A.; Fidalgo, F. Plants Facing Oxidative Challenges—A Little Help from the Antioxidant Networks. Environ. Exp. Bot. 2019, 161, 4–25.
- Singh, R.; Singh, S.; Parihar, P.; Mishra, R.K.; Tripathi, D.K.; Singh, V.P.; Chauhan, D.K.; Prasad, S.M. Reactive Oxygen Species (ROS): Beneficial Companions of Plants’ Developmental Processes. Front. Plant. Sci. 2016, 7, 1299.
- Del Río, L.A. ROS and RNS in Plant Physiology: An Overview. J. Exp. Bot. 2015, 66, 2827–2837.
- Sun, Y.; Lu, Y.; Saredy, J.; Wang, X.; Drummer Iv, C.; Shao, Y.; Saaoud, F.; Xu, K.; Liu, M.; Yang, W.Y.; et al. ROS Systems Are a New Integrated Network for Sensing Homeostasis and Alarming Stresses in Organelle Metabolic Processes. Redox Biol. 2020, 37, 101696.
- Hasanuzzaman, M.; Bhuyan, M.H.M.B.; Parvin, K.; Bhuiyan, T.F.; Anee, T.I.; Nahar, K.; Hossen, M.S.; Zulfiqar, F.; Alam, M.M.; Fujita, M. Regulation of ROS Metabolism in Plants under Environmental Stress: A Review of Recent Experimental Evidence. Int. J. Mol. Sci. 2020, 21, 8695.
- Mittler, R. ROS Are Good. Trends Plant. Sci. 2017, 22, 11–19.
- Ramel, F.; Mialoundama, A.S.; Havaux, M. Nonenzymic Carotenoid Oxidation and Photooxidative Stress Signalling in Plants. J. Exp. Bot. 2013, 64, 799–805.
- Hernández, I.; Alegre, L.; Van Breusegem, F.; Munné-Bosch, S. How Relevant Are Flavonoids as Antioxidants in Plants? Trends Plant. Sci. 2009, 14, 125–132.
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An Overview. J. Nutr. Sci. 2016, 5, e47.
- Dumanović, J.; Nepovimova, E.; Natić, M.; Kuča, K.; Jaćević, V. The Significance of Reactive Oxygen Species and Antioxidant Defense System in Plants: A Concise Overview. Front. Plant. Sci. 2020, 11, 552969.
- Ivanova, A.; Gerasimova, E.; Gazizullina, E. Study of Antioxidant Properties of Agents from the Perspective of Their Action Mechanisms. Molecules 2020, 25, 4251.
- Gulcin, İ. Antioxidants and Antioxidant Methods: An Updated Overview. Arch. Toxicol. 2020, 94, 651–715.
- Munteanu, I.G.; Apetrei, C. Analytical Methods Used in Determining Antioxidant Activity: A Review. Int. J. Mol. Sci. 2021, 22, 3380.
- Pompella, A.; Sies, H.; Wacker, R.; Brouns, F.; Grune, T.; Biesalski, H.K.; Frank, J. The Use of Total Antioxidant Capacity as Surrogate Marker for Food Quality and its Effect on Health is to be Discouraged. Nutrition 2014, 30, 791–793.
- Soccio, M.; Laus, M.N.; Alfarano, M.; Pastore, D. The Soybean Lipoxygenase-Fluorescein Reaction may be used to Assess Antioxidant Capacity of Phytochemicals and Serum. Anal. Methods 2016, 8, 4354–4362.
- Soccio, M.; Laus, M.N.; Alfarano, M.; Dalfino, G.; Panunzio, M.F.; Pastore, D. Antioxidant/Oxidant Balance as a novel approach to evaluate the effect on serum of long-term intake of plant antioxidant-rich foods. J. Funct. Foods 2018, 40, 778–784.
- Franco, R.; Navarro, G.; Martínez-Pinilla, E. Antioxidants versus Food Antioxidant Additives and Food Preservatives. Antioxidants 2019, 8, 542.
- Swann, J.D.; Acosta, D. Failure of Gentamicin to Elevate Cellular Malondialdehyde Content or Increase Generation of Intracellular Reactive Oxygen Species in Primary Cultures of Renal Cortical Epithelial Cells. Biochem. Pharm. 1990, 40, 1523–1526.
- Rosenkranz, A.R.; Schmaldienst, S.; Stuhlmeier, K.M.; Chen, W.; Knapp, W.; Zlabinger, G.J. A Microplate Assay for the Detection of Oxidative Products Using 2′,7′-Dichlorofluorescin-Diacetate. J. Immunol. Methods 1992, 156, 39–45.
- Wang, H.; Joseph, J.A. Quantifying Cellular Oxidative Stress by Dichlorofluorescein Assay Using Microplate Reader. Free Radic. Biol. Med. 1999, 27, 612–616.
- Wolfe, K.L.; Liu, R.H. Cellular Antioxidant Activity (CAA) Assay for Assessing Antioxidants, Foods, and Dietary Supplements. J. Agric. Food Chem. 2007, 55, 8896–8907.
- Wolfe, K.L.; Kang, X.; He, X.; Dong, M.; Zhang, Q.; Liu, R.H. Cellular Antioxidant Activity of Common Fruits. J. Agric. Food Chem. 2008, 56, 8418–8426.
- De la Haba, C.; Palacio, J.R.; Martínez, P.; Morros, A. Effect of Oxidative Stress on Plasma Membrane Fluidity of THP-1 Induced Macrophages. Biochim. Biophys. Acta BBA Biomembr. 2013, 1828, 357–364.
- Park, J.E.; Yang, J.-H.; Yoon, S.J.; Lee, J.-H.; Yang, E.S.; Park, J.-W. Lipid Peroxidation-Mediated Cytotoxicity and DNA Damage in U937 Cells. Biochimie 2002, 84, 1199–1205.
- Sunitha, D. A Review on Antioxidant Methods. Asian J. Pharm. Clin. Res. 2016, 9, 14–32.
- Kellett, M.E.; Greenspan, P.; Pegg, R.B. Modification of the Cellular Antioxidant Activity (CAA) Assay to Study Phenolic Antioxidants in a Caco-2 Cell Line. Food Chem. 2018, 244, 359–363.
- Swift, L.M.; Sarvazyan, N. Localization of Dichlorofluorescin in Cardiac Myocytes: Implications for Assessment of Oxidative Stress. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H982–H990.
- Afri, M.; Frimer, A.A.; Cohen, Y. Active Oxygen Chemistry within the Liposomal Bilayer. Part IV: Locating 2′,7′-Dichlorofluorescein (DCF), 2′,7′-Dichlorodihydrofluorescein (DCFH) and 2′,7′-Dichlorodihydrofluorescein Diacetate (DCFH-DA) in the Lipid Bilayer. Chem. Phys. Lipids 2004, 131, 123–133.
- González, E.; Vaillant, F.; Rojas, G.; Pérez, A. Novel Semiautomated Method for Assessing in Vitro Cellular Antioxidant Activity Using the Light-Scattering Properties of Human Erythrocytes. J. Agric. Food Chem. 2010, 58, 1455–1461.
- González, E.; Vaillant, F.; Pérez, A. In Vitro Cell-Mediated Antioxidant Protection of Human Erythrocytes by Some Common Tropical Fruits. J. Nutr. Food Sci. 2012, 2.
- Dobiasová, S.; Řehořová, K.; Kučerová, D.; Biedermann, D.; Káňová, K.; Petrásková, L.; Koucká, K.; Václavíková, R.; Valentová, K.; Ruml, T.; et al. Multidrug Resistance Modulation Activity of Silybin Derivatives and Their Anti-Inflammatory Potential. Antioxidants 2020, 9, 455.
- Gong, E.S.; Liu, C.; Li, B.; Zhou, W.; Chen, H.; Li, T.; Wu, J.; Zeng, Z.; Wang, Y.; Si, X.; et al. Phytochemical Profiles of Rice and Their Cellular Antioxidant Activity against ABAP Induced Oxidative Stress in Human Hepatocellular Carcinoma HepG2 Cells. Food Chem. 2020, 318, 126484.
- Nizioł-Łukaszewska, Z.; Furman-Toczek, D.; Zagórska-Dziok, M. Antioxidant Activity and Cytotoxicity of Jerusalem Artichoke Tubers and Leaves Extract on HaCaT and BJ Fibroblast Cells. Lipids Health Dis. 2018, 17, 280.
- Royall, J.A.; Ischiropoulos, H. Evaluation of 2′,7′-Dichlorofluorescin and Dihydrorhodamine 123 as Fluorescent Probes for Intracellular H2O2 in Cultured Endothelial Cells. Arch. Biochem. Biophys. 1993, 302, 348–355.
- Yazdani, M. Concerns in the Application of Fluorescent Probes DCDHF-DA, DHR 123 and DHE to Measure Reactive Oxygen Species in Vitro. Toxicol. Vitr. 2015, 30, 578–582.
- Zhang, D.; Xie, L.; Wei, Y.; Liu, Y.; Jia, G.; Zhou, F.; Ji, B. Development of a Cell-Based Antioxidant Activity Assay Using Dietary Fatty Acid as Oxidative Stressor. Food Chem. 2013, 141, 347–356.
- Chen, X.; Zhong, Z.; Xu, Z.; Chen, L.; Wang, Y. 2′,7′-Dichlorodihydrofluorescein as a Fluorescent Probe for Reactive Oxygen Species Measurement: Forty Years of Application and Controversy. Free Radic. Res. 2010, 44, 587–604.
- Quintanar-Escorza, M.A.; González-Martínez, M.T.; del Pilar, I.-O.M.; Calderón-Salinas, J.V. Oxidative Damage Increases Intracellular Free Calcium [Ca2+]i Concentration in Human Erythrocytes Incubated with Lead. Toxicol. In Vitro 2010, 24, 1338–1346.
- Zhou, J.; Gao, G.; Zhang, S.; Wang, H.; Ke, L.; Zhou, J.; Rao, P.; Wang, Q.; Li, J. Influences of Calcium and Magnesium Ions on Cellular Antioxidant Activity (CAA) Determination. Food Chem. 2020, 320, 126625.
- Lubitz, I.; Zikich, D.; Kotlyar, A. Specific High-Affinity Binding of Thiazole Orange to Triplex and G-Quadruplex DNA. Biochemistry 2010, 49, 3567–3574.
- Karunakaran, V.; Pérez Lustres, J.L.; Zhao, L.; Ernsting, N.P.; Seitz, O. Large Dynamic Stokes Shift of DNA Intercalation Dye Thiazole Orange Has Contribution from a High-Frequency Mode. J. Am. Chem. Soc. 2006, 128, 2954–2962.
- Kessel, D. Photodynamic Therapy: A Brief History. J. Clin. Med. 2019, 8, 1581.
- Nygren, J.; Svanvik, N.; Kubista, M. The Interactions between the Fluorescent Dye Thiazole Orange and DNA. Biopolymers 1998, 46, 39–51.
- Armitage, B.A. Cyanine Dye–Nucleic Acid Interactions. In Heterocyclic Polymethine Dyes; Strekowski, L., Ed.; Topics in Heterocyclic Chemistry; Springer: Berlin/Heidelberg, Germany, 2008; Volume 14, pp. 11–29. ISBN 978-3-540-79063-1.
- Derick, S.; Gironde, C.; Perio, P.; Reybier, K.; Nepveu, F.; Jauneau, A.; Furger, C. LUCS (Light-Up Cell System), a Universal High Throughput Assay for Homeostasis Evaluation in Live Cells. Sci. Rep. 2017, 7, 18069.
- Gironde, C.; Rigal, M.; Dufour, C.; Furger, C. AOP1, a New Live Cell Assay for the Direct and Quantitative Measure of Intracellular Antioxidant Effects. Antioxidants 2020, 9, 471.
- Vigliante, I.; Mannino, G.; Maffei, M.E. OxiCyan®, a Phytocomplex of Bilberry (Vaccinium Myrtillus) and Spirulina (Spirulina Platensis), Exerts Both Direct Antioxidant Activity and Modulation of ARE/Nrf2 Pathway in HepG2 Cells. J. Funct. Foods 2019, 61, 103508.
- Bugaj, L.J.; Lim, W.A. High-Throughput Multicolor Optogenetics in Microwell Plates. Nat. Protoc. 2019, 14, 2205–2228.
- Thomas, O.S.; Hörner, M.; Weber, W. A Graphical User Interface to Design High-Throughput Optogenetic Experiments with the OptoPlate-96. Nat. Protoc. 2020, 15, 2785–2787.

