 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maria Isabel Giraldo Giraldo | + 3754 word(s) | 3754 | 2021-04-12 14:28:01 | | | |
| 2 | Peter Tang | Meta information modification | 3754 | 2021-06-16 05:05:40 | | |
Video Upload Options
Ubiquitination of proteins is a post-translational modification process with many different cellular functions, including protein stability, immune signaling, antiviral functions and virus replication. While ubiquitination of viral proteins can be used by the host as a defense mechanism by destroying the incoming pathogen, viruses have adapted to take advantage of this cellular process. The ubiquitin system can be hijacked by viruses to enhance various steps of the replication cycle and increase pathogenesis. Emerging viruses, including severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), flaviviruses like Zika and dengue, as well as highly pathogenic viruses like Ebola and Nipah, have the ability to directly use the ubiquitination process to enhance their viral-replication cycle, and evade immune responses. Some of these mechanisms are conserved among different virus families, especially early during virus entry, providing an opportunity to develop broad-spectrum antivirals.
1. Introduction
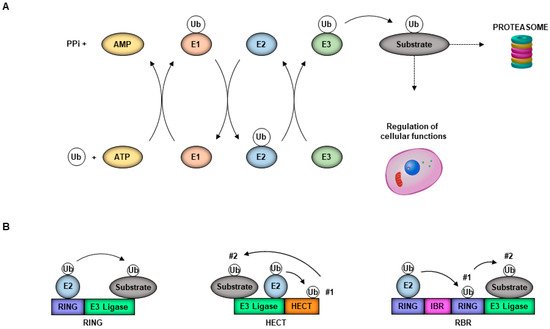
Ubiquitin is a 76-amino acid protein well known for its function in marking proteins for degradation by the proteasome, although multiple non-degradative functions are well established. Covalent ubiquitination depends on a series of enzymes, including the E1 ubiquitin-activating enzyme, an E2 ubiquitin-conjugating enzyme, and an E3 ubiquitin ligase [1]. The E1 ubiquitin-activating enzyme activates ubiquitin in an ATP-dependent fashion [2]. The E2 ubiquitin-conjugating enzyme then forms a complex with the E1. Once the E1 is charged with ubiquitin, the ubiquitin is then transferred to a Cystine on the active site of the E2 [3]. The complex can then interact with one of three different classes of E3 ubiquitin ligases, the HECT, RBR or RING domain ligases; these then bring in a specific substrate. The HECT and RBR E3 ligases can transfer Ub directly to the substrate via formation of an ubiquitin–thioester intermediary on one of the E3 cysteine residues, while RING-containing E3 ligases position the Ub-loaded E2 in proximity to the substrate for E2-to-subtrate transfer, which usually occurs on a lysine (K) on that substrate (Figure 1A,B) [3]. The E3 ubiquitin ligases are interesting because they are the most diverse of the three enzymes, with more than 600 different E3 ligases [4]. Ubiquitin itself has seven lysines, each of which can also be conjugated to another ubiquitin to form a polyubiquitin chain. The E2 conjugase is an important determinant of the type of polyubiquitin chain that can be formed (specifically K6-, K11-, K27-, K29-, K33-, K48- or K63-linked), and the chain linkage is also important for the specific function. Both the E2 and the E3 ligases can be expressed in a cell-type or tissue-specific manner, or their expression can be induced in response to a specific stimulus [4], making the process of ubiquitination a complex process to study. The tripartite motif (TRIM) superfamily of E3 ligases is of particular interest for its roles in regulating the innate immune response and having direct and indirect antiviral activities, and enhancing virus replication [5].
Viruses have evolved to antagonize the host immune response by interfering with host ubiquitin-dependent signaling pathways, or by hijacking the cellular ubiquitination machinery, to promote viral replication and pathogenesis. With the ongoing pandemic caused by the novel strain of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), it is becoming critical to identify common steps used by different viruses to replicate in host cells that could potentially be targeted with broad-spectrum antivirals. Specifically, ubiquitination of viral proteins is emerging as an important host cellular process that is utilized by multiple viruses to promote their replication.
2. Ubiquitin System and Virus Entry
Viruses gain entry to the host cell through highly specific interactions with cellular surface receptors, although some viruses can also use less-specific mechanisms of entry via interactions with lipids or carbohydrates on the surface of the cell membrane [6]. Following attachment, enveloped viruses undergo fusion with the cell at either the cell surface or endosomal membrane (Figure 2) [7]. The exact process of entry differs across viral families, and some viruses have used ubiquitin to enhance this process. This step is critical for productive virus infection and could provide a common target to inhibit multiple viruses. Indeed, many studies have investigated the use of proteasome inhibitors as potential therapeutics for viral infection. It has been reported that for human immunodeficiency virus (HIV), human astrovirus, herpes simplex virus, SARS and other viruses, the use of proteasome inhibitors negatively affects viral replication in vitro [8][9][10][11]. The proteasome can be used in a proviral fashion; however, the use of proteasome inhibitors is also known to deplete free cellular ubiquitin, which could also play a role in affecting viral replication and other functions that are important to maintain cellular homeostasis [12]. The proteasome inhibitor MG132 was found to inhibit mouse hepatitis virus (MHV), a murine coronavirus, at an early entry step, by promoting accumulation of viral RNA in the endosome and potentially inhibiting its release to the cytoplasm [13]. An example of a virus that hijacks the ubiquitination process in a proteasome-dependent manner to promote entry into cells is the flavivirus Japanese encephalitis virus (JEV) [14]. Proteasome inhibitors did not affect JEV attachment to the cells, or viral RNA translation, but did inhibit internalization at the level of releasing viral ribonucleoproteins (RNPs) from the endosome [14]. Additionally, small interfering RNA (siRNA) used to knock down ubiquitin in HeLa cells suggested that reduction in ubiquitin levels negatively impacted JEV entry; however, whether these effects were direct or indirect is unclear [14]. These results partially resemble observations made with a different flavivirus, dengue virus (DENV). When the activity of the E1-ubiquitin-activating enzyme UBA1 was inhibited with the UBEI 41 (Pyr-41) compound, uncoating of DENV was blocked [15]. Although the DENV capsid is degraded by the proteasome, inhibition of the proteasome does not affect virus uncoating. In this case, inhibition of the ubiquitination process by blocking the E1-activating enzyme resulted in inhibition of translation of viral RNA genome, but not virus internalization. Therefore, a ubiquitination step may be necessary for vRNA uncoating [15].
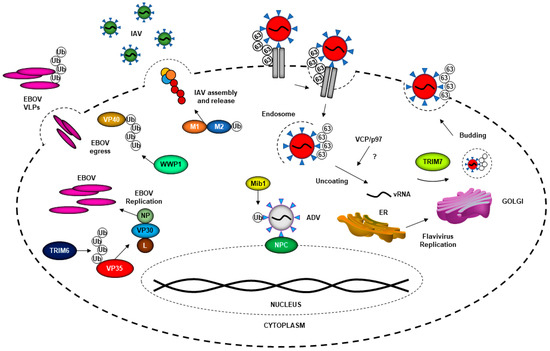
Further evidence that flaviviruses in general may use a non-degradative ubiquitination step for uncoating of the virus genome comes from experiments with yellow fever virus (YFV). Similar to DENV, inhibition of the E1-activating enzyme also inhibited the initial round of YFV translation, and this required the function of the valosin-containing protein VCP/p97 [16], which is known to interact with and extract ubiquitinated proteins from protein complexes [17][18]. Although it is currently unknown which E3-ubiquitin ligase is involved in this process and which viral protein may be ubiquitinated early, these studies strongly support a non-degradative function of ubiquitination of a viral protein that most probably occurs at a post-fusion stage and is required for flavivirus uncoating. However, flaviviruses can also take advantage of the ubiquitination process in an indirect manner. For example, DENV uses the ubiquitination of T cell immunoglobulin mucin 1 (TIM-1) to mediate its entry into cells [19]. On the cytosolic side of TIM-1, two lysine residues (K338 and K346) are needed for TIM-1 ubiquitination [19]. When cells expressing TIM-1 mutants (K338R, K346R and K338/346R) were challenged with DENV, they internalized less virus than when wild type TIM-1 was expressed, indicating that ubiquitination of receptors can also influence DENV entry [19].
While the examples mentioned thus far involve mechanisms in which the intracellular ubiquitination process promotes flavivirus entry, there is evidence that flaviviruses may contain ubiquitinated viral proteins in the infectious virion that promote extracellular interactions with the receptors. For example, a lysine residue (K38) on the envelope (E) protein of Zika virus (ZIKV), which is conserved in other members of the Flaviviridae family (DENV2, West Nile virus (WNV) and YFV), plays a direct role in viral entry [20]. Using recombinant infectious mutant viruses, replication of the ZIKV K38R mutant was significantly attenuated, in a cell-type-specific manner [20]. The host E3-ubiquitin ligase TRIM7 promoted ubiquitination of ZIKV E, and virus replication was reduced, especially in brain and reproductive tissues of infected Trim7−/− animals. A proportion of infectious viral particles released during replication contained ubiquitinated E, and ubiquitination on the E-K38 residue provided the virion the ability to interact with at least one potential cellular receptor, TIM-1, enhancing virus entry, replication and pathogenesis. In this case, ubiquitination of E not only functions in the early steps of virus entry, but also provides a mechanism of tissue tropism [20]. Further evidence that ubiquitination of E promotes better virus attachment and subsequent virus replication came from neutralization experiments using a specific anti-K63-linked-polyubiqutin antibody, which could reduce virus attachment and replication in tissue culture and in vivo [20]. However, the subcellular location where E ubiquitination occurs and how ubiquitinated E is incorporated into the virion remains unknown. An additional ubiquitination unique to ZIKV was on residue K281 of the enveloped protein. Although data suggest that ubiquitination on the E-K281 site may affect a step between virus attachment and uncoating, the precise role of ubiquitination on the K281 site during viral entry remains unclear [20].
Flaviviruses are not the only virus family that can hijack ubiquitin to better enter the cell. Ubiquitination of M1 of influenza A virus (IAV), an orthomyxovirus, has been found to play a role in the release of the virus from the late endosome during entry [21][22]. Human lung adenocarcinoma epithelial cells (A549) treated with shRNA against the E3 ligase ITCH (HECT-type ubiquitin E3 ligase [23]) revealed that there was more viral RNA (vRNA) in the cytoplasm of ITCH knockdown cells, as compared to the control. This inversely correlated with the amount of vRNA in the nucleus, indicating the release of vRNA from endosomes and its transport to the nucleus was delayed [21]. Additional experiments indicated that M1 undergoes direct ubiquitination by ITCH ubiquitin ligase, implicating the role of ubiquitination of M1 in early stages of IAV replication and/or entry [21]. Interestingly, IAV may also use unanchored polyubiquitin chains, which are not covalently attached to any protein, and seemed to be packaged in the infectious virion, for entry and efficient uncoating (Figure 2) [24]. These free ubiquitin chains are recognized by HDAC6, which is a component of the host aggresome pathway [25][26]. Although it is still unclear how IAV packages these unanchored ubiquitin chains, which ubiquitin enzymes are involved in this process, and how this may affect other functions of unanchored ubiquitin, including the innate immune response, this represents additional evidence of multiple ways in which ubiquitin promotes virus internalization and early steps of the replication cycle [27].
Another virus that uses ubiquitin to facilitate entry into cells is adenovirus (ADV). Ubiquitin regulates ADV’s ability to release its genome at the nucleopore of infected cells [28]. It was reported that siRNA-mediated knockdown of the E3-ubiquitin ligase Mind bomb-1 (Mib1) significantly reduced the viral load of ADV infection in vitro, and there was no effect on the early stages of ADV entry [28]. It was also determined that Mib-1 was needed for viral uncoating and genome release (Figure 2) [28].
Ubiquitination and proteasome-dependent degradation of cellular proteins could also provide strategies to limit virus entry. For example, a drug called halofuginone was identified in a screen to induce TMPRSS2 proteasomal degradation via the E3 ubiquitin ligase complex DDB1-CUL4 associated factor DCAF1 [29]. TMPRSS2 is a serine protease that promotes SARS and SARS-CoV-2 entry by proteolytic cleavage of the coronavirus spike protein required for virus attachment to the cell [30]. Proteasome inhibitors have also been proposed to inhibit other steps of the SARS-CoV-2 replication cycle [31].
3. The Ubiquitin System in Promoting Virus Replication
After a virus enters the cell, the virus uses a combination of the host-cell machinery and newly synthetized viral proteins to replicate its viral genome. Viruses have been found to utilize ubiquitin to enhance replication (Figure 2). In several studies, the use of proteasome inhibitors has been shown to block IAV protein synthesis [32][33]. It was discovered that at late stages of the IAV replication cycle, the deubiquitinase (DUB) USP11 can regulate IAV infection in cell-based in vitro assays [33]. Knockdown of USP11 in 293T cells resulted in increased IAV viral titers, while USP11 overexpression decreased viral titers [33]. Based on cellular-localization experiments, USP11 localizes in the nucleus and affects virus replication. Using a catalytic-defective USP11, the authors determined that USP11’s DUB activity is required for IAV RNA replication [33], and that these effects were dependent on monoubiquitination of residue K184 of the viral nucleoprotein (NP) [33]. A mutant K184R remained ubiquitinated because there were multiple ubiquitination sites on NP found by mass spectrometry (MS/MS), and could potentially compensate for the loss of ubiquitination on K184R [34][35]. Primer extension assays showed that ubiquitination is associated with increased transcription, and replication by the IAV polymerase, thus enhancing gene expression during IAV infection [34].
Interestingly, ubiquitination of viral polymerase factors may be a more general mechanism of regulation of the virus RNA transcription and replication steps. For example, the highly pathogenic Ebola virus (EBOV; family Filoviridae) VP35 protein directly uses ubiquitin to facilitate VP35′s polymerase cofactor activity [36]. The host E3-ubiquitin ligase TRIM6 ubiquitinates VP35 at K309 [36]. Although the VP35 K309 ubiquitination site is located in the interferon antagonist domain of VP35, ubiquitination of this residue enhances EBOV replication, and the absence of TRIM6 reduces viral replication (Figure 2) [36]. These results are also supported by experiments using an EBOV minigenome reporter assay, in which overexpression of TRIM6 but not a catalytically inactive mutant (TRIM6-C15A) enhances minigenome luciferase activity [36]. Although the precise mechanism is still unknown, ubiquitination of VP35 may affect interactions with factors of the viral polymerase regulating the balance of virus transcription/replication.
Ubiquitination is also suggested to play a role at the step of virus RNA replication of some flaviviruses. NS1 of DENV is ubiquitinated with K48-linked polyubiquitin chains. The ubiquitination of NS1 K189 was shown to reduce the interaction of NS1 with another viral protein, NS4B [37], and could play a role in the formation of the replication complex during DENV infection. In addition, as mentioned above, proteasome inhibitors reduce the levels of ZIKV and DENV RNA during the late stage of the replication cycle [15][20], suggesting that ubiquitination and subsequent proteasomal degradation of a viral (or host) protein is necessary for efficient virus RNA transcription and/or replication.
4. The Ubiquitin System in Virus Assembly and Budding
The ubiquitin system can be hijacked by viruses, leading to ubiquitination of their viral proteins to allow for assembly and budding. The IAV M2 protein has been shown to play several important roles in the viral life cycle. This protein not only participates in acidification in the endosome to allow uncoating and entry of the virus [38], but it also participates in the late stages of infection, such as assembly, budding and virus release [39]. Recent reports have found that M2 is ubiquitinated and its ubiquitination plays a critical role in the late stage of the influenza virus life cycle [40]. A K78R mutation on M2 reduced its ability to interact with the M1 protein, which is important for the efficient incorporation of vRNP into progeny virions [40]. It was determined that the M2-K78R mutation does not affect IAV entry or replication, but it does affect the assembly or production of infectious virus particles (Figure 2) [40]. The precise mechanism and the specific interaction with the components of the ubiquitin system remain unclear. Lassa virus (LASV), an old-world (OW) arenavirus, is known to cause viral hemorrhagic fever. This virus encodes four proteins: nucleoprotein (NP), surface glycoprotein precursor (GPC), L polymerase and RING finger protein Z. The Z protein plays an important role in viral assembly by assisting in the integration of GP, NP and polymerase L in viral progeny [41]. Z protein interaction with the ITCH E3 ligase was required for LASV replication. This interaction was found to occur through Z protein’s PPXY late domain, as has been shown for a range of other viruses, including lymphocytic choriomeningitis virus (LCMV) [42], and did not require the ubiquitin-E3 ligase activity of ITCH. Furthermore, ITCH is important for the production of infectious LASV particles and necessary for the release of viral progeny [43]. In EBOV, the PPxY late-domain motif of VP40 interacts with the WW domain of ITCH. This interaction regulates the budding of EBOV virus-like particles (VLPs) [44]. EBOV VP40 also has been shown to be SUMOylated, and this may regulate the stability of VP40. It was also proposed that SUMO and possibly ubiquitin could be incorporated into the VLPs, but the functions were not elucidated [45]. A similar mechanism has been reported between EBOV VP40 and the E3 ligase WWP1, in which interactions between the two are required for VLP budding (Figure. 2) [46]. Nedd4 is another E3-ubiquitin ligase that belongs to the HECT-type ubiquitin E3 ligase family [23] and is important in budding for EBOV and marburg virus (MARV) [47][48]. Nedd4 is found in the perinuclear region and can be associated with lipid rafts at the cytoplasmic membrane [48][49]. The protein structure of Nedd4 consists of a C2 membrane-binding domain, four central WW domains that bind to adaptors that generally contain PY motifs (PPxY or LPxY x is any residue) in target proteins [47] and a C-terminus homologous to the E6-AP carboxyl terminus (HECT) ubiquitin ligase domain [50]. Budding of EBOV and MARV VLPs in the presence of Nedd4 is perhaps modulated by Nedd4 ubiquitination, since there is a significant reduction when the HECT domain is mutated compared to wild-type Nedd4 [47][48].
LCMV has been increasingly recognized as a teratogen in recent years [51]. Nedd4 E3 Ub ligase is required for the release of LCMV particles, and this ubiquitination is used as a mechanism for the recruitment of ESCRT mediated by PPXY, an important complex for viral budding, suggesting that ubiquitination can be generated by other Z-associated proteins [42]. Paramyxoviruses can also utilize the ubiquitin system for the nuclear–cytoplasmic trafficking of the matrix protein (M). This was corroborated by mutations in the putative bipartite nuclear localization signal (NLS) and the leucine-rich nuclear export signal (NES) found in Nipah virus (NiV), Hendra virus (HeV), Sendai virus (SeV) and mumps virus [52], which prevented nuclear trafficking and budding. Through the ectopic expression of ubiquitin, an increase in budding was observed, but when proteasome inhibitors such as bortezomib were used, there was nuclear retention of M protein, and viral budding was blocked [52][53].
5. The Ubiquitin System and Viral Evasion of the Type-I Interferon (IFN-I) Response
The innate immune system is the first defense against pathogens and can detect virus invasion to limit virus replication. Innate immunity is activated when pattern recognition receptors (PRRs) that include Toll-like receptors (TLRs) and cytoplasmic RIG-I-like receptors (RLRs), recognize microbial components encoded in microorganisms that are known as pathogen-associated molecular patterns (PAMPs) [54]. Recognition of viral products by PRRs triggers multiple downstream signaling cascades via activation of kinases, including IκB kinases IKKα/β/ε, TBK1, TAK1 and others. Phosphorylation of multiple transcription factors including, but not limited to, IRF3, IRF7 and NF-κB, promotes their translocation to the nucleus, resulting in induction of IFN-I and proinflammatory cytokines [54]. RIG-I has an important role in virus RNA recognition and is activated upon ubiquitination by the E3-ubiquitin ligases TRIM25 and Riplet [55][56], and subsequent downstream signaling via the adaptor protein MAVS, which is also heavily regulated by the ubiquitination process, leading to IFN-I production [57]. Upon release from infected cells, IFNs are recognized by their receptor in an autocrine or paracrine manner and activate the Janus kinase signal transducer and activator of transcription (JAK-STAT) signaling pathway, leading to induction of antiviral host-effector proteins, called IFN-stimulated genes (ISGs) [58]. In addition, the E3-ubiquitin ligase TRIM6 regulates the phosphorylation and activation of IKKε-dependent STAT1 phosphorylation, and this can occur via VAMP8, a membrane protein associated with vesicles [59]. ISGs have a broad scope of antiviral mechanisms that are able to counter the viruses at different stages of their life cycles. Viruses have evolved multiple mechanism to block almost every step of these pathways. In many cases, this includes the inhibition of E3-ubiquitin ligases involved in the signaling, as discussed below (see Figure 3).
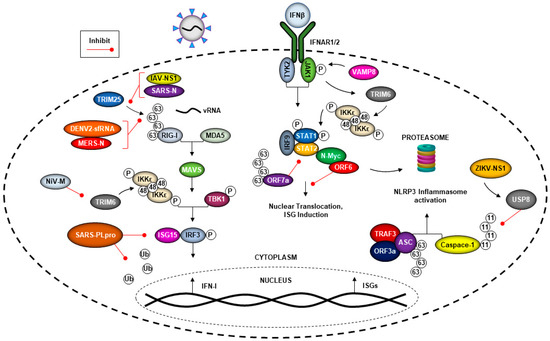
Figure 3. Viruses can evade immune responses utilizing the ubiquitin system. Viruses have evolved the ability to evade the host-innate immune response by antagonizing IFN production and signaling. SARS-CoV ORF6 can interact with N-Myc to promote its degradation through the ubiquitin proteasome, while ORF7a utilizes K63-linked polyubiquitin chains to prevent STAT2 phosphorylation and IFN-I signaling. The N protein of both SARS and MERS-CoV block the interaction between TRIM25 and RIG-I, preventing the K63-linked polyubiquitination of RIG-I needed for IFN-I production. TRIM25 is also a target of inhibition by the NS1 protein of IAV and the short noncoding sfRNAs of dengue-2 (DENV2). The PLpro component of SARS-CoV is capable of antagonizing innate immune pathways by acting as a deubiquitinase and by preventing the ISGylation of cellular proteins, including IRF3. Both SARS-CoV and Zika virus (ZIKV) alter components of the host ubiquitin system to activate the NLRP3 inflammasome, potentially leading to greater dissemination of viral progeny. The NS1 protein of ZIKV can recruit the deubiquitinase USP8 to cleave the K11-linked polyubiquitin chains from Caspase-1, while the SARS-CoV ORF3a protein promotes TRAF3 and ASC association, resulting in the K63-linked ubiquitination of ASC. In addition, ubiquitination also possesses antiviral functions. TRIM6 can regulate the expression of VAMP8 to promote JAK1 phosphorylation downstream of IFN-I signaling and promote the synthesis of unanchored K48-linked polyubiquitin chains for IKKε oligomerization.
References
- Li, X.; Elmira, E.; Rohondia, S.; Wang, J.; Liu, J.; Dou, Q.P. A patent review of the ubiquitin ligase system: 2015–2018. Expert. Opin. Ther. Pat. 2018, 28, 919–937.
- Zhuang, J.; Shirazi, F.; Singh, R.K.; Kuiatse, I.; Wang, H.; Lee, H.C.; Berkova, Z.; Berger, A.; Hyer, M.; Chattopadhyay, N.; et al. Ubiquitin-activating enzyme inhibition induces an unfolded protein response and overcomes drug resistance in myeloma. Blood 2019, 133, 1572–1584.
- Ye, Y.; Rape, M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764.
- Hage, A.; Rajsbaum, R. To TRIM or not to TRIM: The balance of host-virus interactions mediated by the ubiquitin system. J. Gen. Virol. 2019, 100, 1641–1662.
- Giraldo, M.I.; Hage, A.; Van Tol, S.; Rajsbaum, R. TRIM Proteins in Host Defense and Viral Pathogenesis. Curr. Clin. Microbiol. Rep. 2020, 1–14.
- Marsh, M.; Helenius, A. Virus entry: Open sesame. Cell 2006, 124, 729–740.
- Cohen, F.S. How Viruses Invade Cells. Biophys. J. 2016, 110, 1028–1032.
- Yu, L.; Mohanram, V.; Simonson, O.E.; Smith, C.I.; Spetz, A.L.; Mohamed, A.J. Proteasome inhibitors block HIV-1 replication by affecting both cellular and viral targets. Biochem. Biophys. Res. Commun. 2009, 385, 100–105.
- Casorla-Pérez, L.A.; López, T.; López, S.; Arias, C.F. The Ubiquitin-Proteasome System Is Necessary for Efficient Replication of Human Astrovirus. J. Virol. 2018, 92.
- Schneider, S.M.; Pritchard, S.M.; Wudiri, G.A.; Trammell, C.E.; Nicola, A.V. Early Steps in Herpes Simplex Virus Infection Blocked by a Proteasome Inhibitor. mBio 2019, 10.
- Schneider, M.; Ackermann, K.; Stuart, M.; Wex, C.; Protzer, U.; Schätzl, H.M.; Gilch, S. Severe acute respiratory syndrome coronavirus replication is severely impaired by MG132 due to proteasome-independent inhibition of M-calpain. J. Virol. 2012, 86, 10112–10122.
- Schneider, S.M.; Lee, B.H.; Nicola, A.V. Viral entry and the ubiquitin-proteasome system. Cell. Microbiol. 2020.
- Yu, G.Y.; Lai, M.M. The ubiquitin-proteasome system facilitates the transfer of murine coronavirus from endosome to cytoplasm during virus entry. J. Virol. 2005, 79, 644–648.
- Wang, S.; Liu, H.; Zu, X.; Liu, Y.; Chen, L.; Zhu, X.; Zhang, L.; Zhou, Z.; Xiao, G.; Wang, W. The ubiquitin-proteasome system is essential for the productive entry of Japanese encephalitis virus. Virology 2016, 498, 116–127.
- Byk, L.A.; Iglesias, N.G.; De Maio, F.A.; Gebhard, L.G.; Rossi, M.; Gamarnik, A.V. Dengue Virus Genome Uncoating Requires Ubiquitination. mBio 2016, 7.
- Ramanathan, H.N.; Zhang, S.; Douam, F.; Mar, K.B.; Chang, J.; Yang, P.L.; Schoggins, J.W.; Ploss, A.; Lindenbach, B.D. A Sensitive Yellow Fever Virus Entry Reporter Identifies Valosin-Containing Protein (VCP/p97) as an Essential Host Factor for Flavivirus Uncoating. mBio 2020, 11.
- Van den Boom, J.; Meyer, H. VCP/p97-Mediated Unfolding as a Principle in Protein Homeostasis and Signaling. Mol. Cell 2018, 69, 182–194.
- Ye, Y.; Tang, W.K.; Zhang, T.; Xia, D. A Mighty "Protein Extractor" of the Cell: Structure and Function of the p97/CDC48 ATPase. Front. Mol. Biosci. 2017, 4, 39.
- Dejarnac, O.; Hafirassou, M.L.; Chazal, M.; Versapuech, M.; Gaillard, J.; Perera-Lecoin, M.; Umana-Diaz, C.; Bonnet-Madin, L.; Carnec, X.; Tinevez, J.Y.; et al. TIM-1 Ubiquitination Mediates Dengue Virus Entry. Cell Rep. 2018, 23, 1779–1793.
- Giraldo, M.I.; Xia, H.; Aguilera-Aguirre, L.; Hage, A.; Van Tol, S.; Shan, C.; Xie, X.; Sturdevant, G.L.; Robertson, S.J.; McNally, K.L.; et al. Envelope protein ubiquitination drives entry and pathogenesis of Zika virus. Nature 2020, 585, 414–419.
- Su, W.C.; Chen, Y.C.; Tseng, C.H.; Hsu, P.W.; Tung, K.F.; Jeng, K.S.; Lai, M.M. Pooled RNAi screen identifies ubiquitin ligase Itch as crucial for influenza A virus release from the endosome during virus entry. Proc. Natl. Acad. Sci. USA 2013, 110, 17516–17521.
- Rudnicka, A.; Yamauchi, Y. Ubiquitin in Influenza Virus Entry and Innate Immunity. Viruses 2016, 8, 293.
- Sluimer, J.; Distel, B. Regulating the human HECT E3 ligases. Cell Mol. Life Sci. 2018, 75, 3121–3141.
- Banerjee, I.; Miyake, Y.; Nobs, S.P.; Schneider, C.; Horvath, P.; Kopf, M.; Matthias, P.; Helenius, A.; Yamauchi, Y. Influenza A virus uses the aggresome processing machinery for host cell entry. Science 2014, 346, 473–477.
- Hao, R.; Nanduri, P.; Rao, Y.; Panichelli, R.S.; Ito, A.; Yoshida, M.; Yao, T.P. Proteasomes activate aggresome disassembly and clearance by producing unanchored ubiquitin chains. Mol. Cell 2013, 51, 819–828.
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738.
- Rajsbaum, R.; García-Sastre, A. Virology. Unanchored ubiquitin in virus uncoating. Science 2014, 346, 427–428.
- Bauer, M.; Flatt, J.W.; Seiler, D.; Cardel, B.; Emmenlauer, M.; Boucke, K.; Suomalainen, M.; Hemmi, S.; Greber, U.F. The E3 Ubiquitin Ligase Mind Bomb 1 Controls Adenovirus Genome Release at the Nuclear Pore Complex. Cell Rep. 2019, 29, 3785–3795.
- Chen, Y.; Lear, T.; Evankovich, J.; Larsen, M.; Lin, B.; Alfaras, I.; Kennerdell, J.; Salminen, L.; Camarco, D.; Lockwood, K.; et al. A high throughput screen for TMPRSS2 expression identifies FDA-approved and clinically advanced compounds that can limit SARS-CoV-2 entry. Res. Sq. 2020.
- Iwata-Yoshikawa, N.; Okamura, T.; Shimizu, Y.; Hasegawa, H.; Takeda, M.; Nagata, N. TMPRSS2 Contributes to Virus Spread and Immunopathology in the Airways of Murine Models after Coronavirus Infection. J. Virol. 2019, 93.
- Longhitano, L.; Tibullo, D.; Giallongo, C.; Lazzarino, G.; Tartaglia, N.; Galimberti, S.; Li Volti, G.; Palumbo, G.A.; Liso, A. Proteasome Inhibitors as a Possible Therapy for SARS-CoV-2. Int. J. Mol. Sci. 2020, 21, 3622.
- Rodriguez, A.; Pérez-González, A.; Nieto, A. Influenza virus infection causes specific degradation of the largest subunit of cellular RNA polymerase II. J. Virol. 2007, 81, 5315–5324.
- Liao, T.L.; Wu, C.Y.; Su, W.C.; Jeng, K.S.; Lai, M.M. Ubiquitination and deubiquitination of NP protein regulates influenza A virus RNA replication. EMBO J. 2010, 29, 3879–3890.
- Kirui, J.; Mondal, A.; Mehle, A. Ubiquitination Upregulates Influenza Virus Polymerase Function. J. Virol. 2016, 90, 10906–10914.
- Lin, Y.C.; Jeng, K.S.; Lai, M.M.C. CNOT4-Mediated Ubiquitination of Influenza A Virus Nucleoprotein Promotes Viral RNA Replication. mBio 2017, 8.
- Bharaj, P.; Atkins, C.; Luthra, P.; Giraldo, M.I.; Dawes, B.E.; Miorin, L.; Johnson, J.R.; Krogan, N.J.; Basler, C.F.; Freiberg, A.N.; et al. The Host E3-Ubiquitin Ligase TRIM6 Ubiquitinates the Ebola Virus VP35 Protein and Promotes Virus Replication. J. Virol. 2017, 91.
- Giraldo, M.I.; Vargas-Cuartas, O.; Gallego-Gomez, J.C.; Shi, P.Y.; Padilla-Sanabria, L.; Castaño-Osorio, J.C.; Rajsbaum, R. K48-linked polyubiquitination of dengue virus NS1 protein inhibits its interaction with the viral partner NS4B. Virus Res. 2018, 246, 1–11.
- Pinto, L.H.; Holsinger, L.J.; Lamb, R.A. Influenza virus M2 protein has ion channel activity. Cell 1992, 69, 517–528.
- Rossman, J.S.; Jing, X.; Leser, G.P.; Lamb, R.A. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 2010, 142, 902–913.
- Su, W.C.; Yu, W.Y.; Huang, S.H.; Lai, M.M.C. Ubiquitination of the Cytoplasmic Domain of Influenza A Virus M2 Protein Is Crucial for Production of Infectious Virus Particles. J. Virol. 2018, 92.
- Stott, R.J.; Strecker, T.; Foster, T.L. Distinct Molecular Mechanisms of Host Immune Response Modulation by Arenavirus NP and Z Proteins. Viruses 2020, 12, 784.
- Ziegler, C.M.; Dang, L.; Eisenhauer, P.; Kelly, J.A.; King, B.R.; Klaus, J.P.; Manuelyan, I.; Mattice, E.B.; Shirley, D.J.; Weir, M.E.; et al. NEDD4 family ubiquitin ligases associate with LCMV Z’s PPXY domain and are required for virus budding, but not via direct ubiquitination of Z. PLoS Pathog. 2019, 15.
- Baillet, N.; Krieger, S.; Carnec, X.; Mateo, M.; Journeaux, A.; Merabet, O.; Caro, V.; Tangy, F.; Vidalain, P.O.; Baize, S. E3 Ligase ITCH Interacts with the Z Matrix Protein of Lassa and Mopeia Viruses and Is Required for the Release of Infectious Particles. Viruses 2019, 12, 49.
- Han, Z.; Sagum, C.A.; Bedford, M.T.; Sidhu, S.S.; Sudol, M.; Harty, R.N. ITCH E3 Ubiquitin Ligase Interacts with Ebola Virus VP40 To Regulate Budding. J. Virol. 2016, 90, 9163–9171.
- Baz-Martinez, M.; El Motiam, A.; Ruibal, P.; Condezo, G.N.; De la Cruz-Herrera, C.F.; Lang, V.; Collado, M.; San Martin, C.; Rodriguez, M.S.; Munoz-Fontela, C.; et al. Regulation of Ebola virus VP40 matrix protein by SUMO. Sci. Rep. 2016, 6, 37258.
- Han, Z.; Sagum, C.A.; Takizawa, F.; Ruthel, G.; Berry, C.T.; Kong, J.; Sunyer, J.O.; Freedman, B.D.; Bedford, M.T.; Sidhu, S.S.; et al. Ubiquitin Ligase WWP1 Interacts with Ebola Virus VP40 To Regulate Egress. J. Virol. 2017, 91.
- Yasuda, J.; Nakao, M.; Kawaoka, Y.; Shida, H. Nedd4 regulates egress of Ebola virus-like particles from host cells. J. Virol. 2003, 77, 9987–9992.
- Urata, S.; Yasuda, J. Regulation of Marburg virus (MARV) budding by Nedd4.1: A different WW domain of Nedd4.1 is critical for binding to MARV and Ebola virus VP40. J. Gen. Virol. 2010, 91, 228–234.
- Lafont, F.; Simons, K. Raft-partitioning of the ubiquitin ligases Cbl and Nedd4 upon IgE-triggered cell signaling. Proc. Natl. Acad. Sci. USA 2001, 98, 3180–3184.
- Harvey, K.F.; Kumar, S. Nedd4-like proteins: An emerging family of ubiquitin-protein ligases implicated in diverse cellular functions. Trends Cell Biol. 1999, 9, 166–169.
- Bonthius, D.J.; Perlman, S. Congenital viral infections of the brain: Lessons learned from lymphocytic choriomeningitis virus in the neonatal rat. PLoS Pathog. 2007, 3, 149.
- Pentecost, M.; Vashisht, A.A.; Lester, T.; Voros, T.; Beaty, S.M.; Park, A.; Wang, Y.E.; Yun, T.E.; Freiberg, A.N.; Wohlschlegel, J.A.; et al. Evidence for ubiquitin-regulated nuclear and subnuclear trafficking among Paramyxovirinae matrix proteins. PLoS Pathog. 2015, 11, 1004739.
- Wang, Y.E.; Park, A.; Lake, M.; Pentecost, M.; Torres, B.; Yun, T.E.; Wolf, M.C.; Holbrook, M.R.; Freiberg, A.N.; Lee, B. Ubiquitin-regulated nuclear-cytoplasmic trafficking of the Nipah virus matrix protein is important for viral budding. PLoS Pathog. 2010, 6, 1001186.
- Hoffmann, J.; Akira, S. Innate immunity. Curr. Opin. Immunol. 2013, 25, 1–3.
- Oshiumi, H.; Miyashita, M.; Inoue, N.; Okabe, M.; Matsumoto, M.; Seya, T. The ubiquitin ligase Riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection. Cell Host Microbe 2010, 8, 496–509.
- Oshiumi, H. Recent Advances and Contradictions in the Study of the Individual Roles of Ubiquitin Ligases That Regulate RIG-I-Like Receptor-Mediated Antiviral Innate Immune Responses. Front. Immunol. 2020, 11, 1296.
- Liu, B.; Gao, C. Regulation of MAVS activation through post-translational modifications. Curr. Opin. Immunol. 2018, 50, 75–81.
- Iwasaki, A. A virological view of innate immune recognition. Annu. Rev. Microbiol. 2012, 66, 177–196.
- Van Tol, S.; Atkins, C.; Bharaj, P.; Johnson, K.N.; Hage, A.; Freiberg, A.N.; Rajsbaum, R. VAMP8 Contributes to the TRIM6-Mediated Type I Interferon Antiviral Response during West Nile Virus Infection. J. Virol. 2020, 94.

