 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sheng-Han Wang | + 4240 word(s) | 4240 | 2021-06-03 06:03:15 | | | |
| 2 | Lily Guo | Meta information modification | 4240 | 2021-06-15 03:11:00 | | |
Video Upload Options
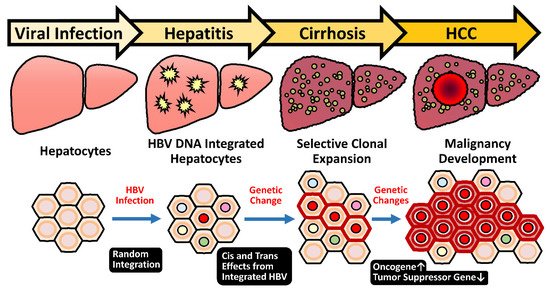
Hepatitis B virus (HBV) infection is the major risk factor for hepatocellular carcinoma (HCC). Understanding the unique features for HBV-induced HCC can shed new light on the unmet needs in its early diagnosis and effective therapy. During decades of chronic hepatitis B, hepatocytes undergoing repeated damage and regeneration accumulate genetic changes predisposing to HCC development. In addition to traditional mutations in viral and cellular oncogenes, HBV integration into the cell chromosomes is an alternative genetic change contributing to hepatocarcinogenesis. A striking male dominance in HBV-related HCC further highlights an interaction between androgen sex hormone and viral factors, which contributes to the gender difference via stimulating viral replication and activation of oncogenes preferentially in male patients. Meanwhile, a novel circulating tumor biomarker generated by HBV integration shows great potential for the early diagnosis of HCC. These unique HBV-induced hepatocarcinogenic mechanisms provide new insights for the future development of superior diagnosis and treatment strategies.
1. Introduction
2. Pathogenesis of HBV-Related HCC
2.1. Chronic Inflammation and Hepatocyte Regeneration
2.2. HBV Genotypes and Specific HBV Variants
2.3. HBx: A Multifunctional Viral Protein with Versatile Oncogenic Activities
2.3.1. HBx Stimulates HBV Gene Expression
2.3.2. HBx Effects on Tumor-Related Characteristics
2.3.3. HBx Mutants and HCC Development
2.4. Sex Hormones in Regulating the Gender Difference of Carcinogenesis
2.4.1. The HBx–AR Circuit in Promoting Male HCC
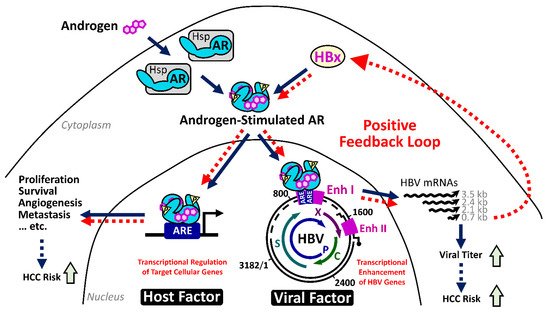
2.4.2. Estrogen/Estrogen Receptor Pathway in Suppressing Female HCC
2.5. HBV Integration Induced Mutagenesis and Genomic Instability
2.5.1. Random Integration of HBV with Selective Hotspots in HCC Genome
2.5.2. HBV-Induced Insertional Mutagenesis Is Responsive to Sex Hormone Regulation
2.5.3. Genome Instability Caused by HBV Integration
2.6. Heavy Alcohol Consumption and Risk of HBV-Related Disease Progression
References
- Brar, G.; Greten, T.F.; Graubard, B.I.; McNeel, T.S.; Petrick, J.L.; McGlynn, K.A.; Altekruse, S.F. Hepatocellular carcinoma survival by etiology: A SEER-medicare database analysis. Hepatol. Commun. 2020, 4, 1541–1551.
- Petruzziello, A. Epidemiology of hepatitis B virus (HBV) and hepatitis C virus (HCV) related hepatocellular carcinoma. Open Virol. J. 2018, 12, 26–32.
- Park, N.H.; Chung, Y.H.; Lee, H.S. Impacts of vaccination on hepatitis B viral infections in Korea over a 25-year period. Intervirology 2010, 53, 20–28.
- Luo, Z.; Li, L.; Ruan, B. Impact of the implementation of a vaccination strategy on hepatitis B virus infections in China over a 20-year period. Int. J. Infect. Dis. 2012, 16, e82–e88.
- Chen, D.S.; Hsu, N.H.; Sung, J.L.; Hsu, T.C.; Hsu, S.T.; Kuo, Y.T.; Lo, K.J.; Shih, Y.T. A mass vaccination program in Taiwan against hepatitis B virus infection in infants of hepatitis B surface antigen-carrier mothers. JAMA J. Am. Med. Assoc. 1987, 257, 2597–2603.
- Chang, M.H.; Chen, C.J.; Lai, M.S.; Hsu, H.M.; Wu, T.C.; Kong, M.S.; Liang, D.C.; Shau, W.Y.; Chen, D.S. Universal hepatitis B vaccination in Taiwan and the incidence of hepatocellular carcinoma in children. Taiwan Childhood Hepatoma Study Group. N. Engl. J. Med. 1997, 336, 1855–1859.
- Ni, Y.H.; Chen, D.S. Hepatitis B vaccination in children: The Taiwan experience. Pathol. Biol. 2010, 58, 296–300.
- Polaris Observatory. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: A modelling study. Lancet Gastroenterol. Hepatol. 2018, 3, 383–403.
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101.
- Lavanchy, D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J. Viral Hepat. 2004, 11, 97–107.
- Liaw, Y.F. Hepatitis B virus replication and liver disease progression: The impact of antiviral therapy. Antivir. Ther. 2006, 11, 669–679.
- Caviglia, G.P.; Abate, M.L.; Pellicano, R.; Smedile, A. Chronic hepatitis B therapy: Available drugs and treatment guidelines. Minerva Gastroenterol. Dietol. 2015, 61, 61–70.
- Choi, W.M.; Choi, J.; Lim, Y.S. Effects of tenofovir vs. entecavir on risk of hepatocellular carcinoma in patients with chronic HBV infection: A systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2021, 19, 246–258.e9.
- Yip, T.C.; Lai, J.C.; Wong, G.L. Secondary prevention for hepatocellular carcinoma in patients with chronic hepatitis B: Are all the nucleos(t)ide analogues the same? J. Gastroenterol. 2020, 55, 1023–1036.
- Tseng, C.H.; Tseng, C.M.; Wu, J.L.; Hsu, Y.C.; El-Serag, H.B. Magnitude of and prediction for risk of hepatocellular carcinoma in patients with chronic hepatitis B taking entecavir or tenofovir therapy: A systematic review. J. Gastroenterol. Hepatol. 2020, 35, 1684–1693.
- Saitta, C.; Tripodi, G.; Barbera, A.; Bertuccio, A.; Smedile, A.; Ciancio, A.; Raffa, G.; Sangiovanni, A.; Navarra, G.; Raimondo, G.; et al. Hepatitis B virus (HBV) DNA integration in patients with occult HBV infection and hepatocellular carcinoma. Liver Int. 2015, 35, 2311–2317.
- Summers, J.; Jilbert, A.R.; Yang, W.; Aldrich, C.E.; Saputelli, J.; Litwin, S.; Toll, E.; Mason, W.S. Hepatocyte turnover during resolution of a transient hepadnaviral infection. Proc. Natl. Acad. Sci. USA 2003, 100, 11652–11659.
- Tu, T.; Budzinska, M.A.; Vondran, F.W.R.; Shackel, N.A.; Urban, S. Hepatitis B virus DNA integration occurs early in the viral life cycle in an in vitro infection model via sodium taurocholate cotransporting polypeptide-dependent uptake of enveloped virus particles. J. Virol. 2018, 92.
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769.
- Akcay, I.M.; Katrinli, S.; Ozdil, K.; Doganay, G.D.; Doganay, L. Host genetic factors affecting hepatitis B infection outcomes: Insights from genome-wide association studies. World J. Gastroenterol. 2018, 24, 3347–3360.
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273.e1.
- Lee, C.M.; Lu, S.N.; Changchien, C.S.; Yeh, C.T.; Hsu, T.T.; Tang, J.H.; Wang, J.H.; Lin, D.Y.; Chen, C.L.; Chen, W.J. Age, gender, and local geographic variations of viral etiology of hepatocellular carcinoma in a hyperendemic area for hepatitis B virus infection. Cancer 1999, 86, 1143–1150.
- Chu, C.M.; Liaw, Y.F.; Sheen, I.S.; Lin, D.Y.; Huang, M.J. Sex difference in chronic hepatitis B virus infection: An appraisal based on the status of hepatitis B antigen and antibody. Hepatology 1983, 3, 947–950.
- Chen, C.J.; Yang, H.I.; Iloeje, U.H. Hepatitis B virus DNA levels and outcomes in chronic hepatitis B. Hepatology 2009, 49, S72–S84.
- Su, F.H.; Chen, J.D.; Cheng, S.H.; Lin, C.H.; Liu, Y.H.; Chu, F.Y. Seroprevalence of hepatitis-B infection amongst Taiwanese university students 18 years following the commencement of a national hepatitis-B vaccination program. J. Med. Virol. 2007, 79, 138–143.
- Liu, C.J.; Chen, P.J. Elimination of hepatitis B in highly endemic settings: Lessons learned in Taiwan and challenges ahead. Viruses 2020, 12, 815.
- Multhoff, G.; Molls, M.; Radons, J. Chronic inflammation in cancer development. Front. Immunol. 2011, 2, 98.
- Arbuthnot, P.; Kew, M. Hepatitis B virus and hepatocellular carcinoma. Int. J. Exp. Pathol. 2001, 82, 77–100.
- Wong, G.L.; Lampertico, P. Residual risk of HCC during long-term oral nucleos(t)ide analogues (NUCs) in patients with CHB —Is one NUC better than the other? J. Hepatol. 2019, 71, 453–455.
- Yip, T.C.; Wong, V.W.; Chan, H.L.; Tse, Y.K.; Lui, G.C.; Wong, G.L. Tenofovir is associated with lower risk of hepatocellular carcinoma than entecavir in patients with chronic HBV infection in China. Gastroenterology 2020, 158, 215–225.e6.
- Thimme, R.; Wieland, S.; Steiger, C.; Ghrayeb, J.; Reimann, K.A.; Purcell, R.H.; Chisari, F.V. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J. Virol. 2003, 77, 68–76.
- Xu, L.; Yin, W.; Sun, R.; Wei, H.; Tian, Z. Kupffer cell-derived IL-10 plays a key role in maintaining humoral immune tolerance in hepatitis B virus-persistent mice. Hepatology 2014, 59, 443–452.
- Li, M.; Sun, R.; Xu, L.; Yin, W.; Chen, Y.; Zheng, X.; Lian, Z.; Wei, H.; Tian, Z. Kupffer cells support hepatitis B virus-mediated CD8+ T cell exhaustion via hepatitis B core antigen-TLR2 interactions in mice. J. Immunol. 2015, 195, 3100–3109.
- Cheng, Y.; Zhu, Y.O.; Becht, E.; Aw, P.; Chen, J.; Poidinger, M.; Sessions, P.F.D.; Hibberd, M.L.; Bertoletti, A.; Lim, S.G.; et al. Multifactorial heterogeneity of virus-specific T cells and association with the progression of human chronic hepatitis B infection. Sci. Immunol. 2019, 4.
- Reignat, S.; Webster, G.J.; Brown, D.; Ogg, G.S.; King, A.; Seneviratne, S.L.; Dusheiko, G.; Williams, R.; Maini, M.K.; Bertoletti, A. Escaping high viral load exhaustion: CD8 cells with altered tetramer binding in chronic hepatitis B virus infection. J. Exp. Med. 2002, 195, 1089–1101.
- Hoogeveen, R.C.; Robidoux, M.P.; Schwarz, T.; Heydmann, L.; Cheney, J.A.; Kvistad, D.; Aneja, J.; Melgaco, J.G.; Fernandes, C.A.; Chung, R.T.; et al. Phenotype and function of HBV-specific T cells is determined by the targeted epitope in addition to the stage of infection. Gut 2019, 68, 893–904.
- Schuch, A.; Alizei, E.S.; Heim, K.; Wieland, D.; Kiraithe, M.M.; Kemming, J.; Llewellyn-Lacey, S.; Sogukpinar, O.; Ni, Y.; Urban, S.; et al. Phenotypic and functional differences of HBV core-specific versus HBV polymerase-specific CD8+ T cells in chronically HBV-infected patients with low viral load. Gut 2019, 68, 905–915.
- Webster, G.J.; Reignat, S.; Brown, D.; Ogg, G.S.; Jones, L.; Seneviratne, S.L.; Williams, R.; Dusheiko, G.; Bertoletti, A. Longitudinal analysis of CD8+ T cells specific for structural and nonstructural hepatitis B virus proteins in patients with chronic hepatitis B: Implications for immunotherapy. J. Virol. 2004, 78, 5707–5719.
- Bertoletti, A.; Kennedy, P.T.F.; Durantel, D. HBV infection and HCC: The ‘dangerous liaisons’. Gut 2018, 67, 787–788.
- Kim, G.A.; Lim, Y.S.; Han, S.; Choi, J.; Shim, J.H.; Kim, K.M.; Lee, H.C.; Lee, Y.S. High risk of hepatocellular carcinoma and death in patients with immune-tolerant-phase chronic hepatitis B. Gut 2018, 67, 945–952.
- Mason, W.S.; Gill, U.S.; Litwin, S.; Zhou, Y.; Peri, S.; Pop, O.; Hong, M.L.; Naik, S.; Quaglia, A.; Bertoletti, A.; et al. HBV DNA integration and clonal hepatocyte expansion in chronic hepatitis B patients considered immune tolerant. Gastroenterology 2016, 151, 986–998 e4.
- Maini, M.K.; Boni, C.; Lee, C.K.; Larrubia, J.R.; Reignat, S.; Ogg, G.S.; King, A.S.; Herberg, J.; Gilson, R.; Alisa, A.; et al. The role of virus-specific CD8(+) cells in liver damage and viral control during persistent hepatitis B virus infection. J. Exp. Med. 2000, 191, 1269–1280.
- Lin, C.L.; Kao, J.H. Natural history of acute and chronic hepatitis B: The role of HBV genotypes and mutants. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 249–255.
- McMahon, B.J. The influence of hepatitis B virus genotype and subgenotype on the natural history of chronic hepatitis B. Hepatol. Int. 2009, 3, 334–342.
- Sunbul, M. Hepatitis B virus genotypes: Global distribution and clinical importance. World J. Gastroenterol. 2014, 20, 5427–5434.
- Kao, J.H.; Chen, P.J.; Lai, M.Y.; Chen, D.S. Basal core promoter mutations of hepatitis B virus increase the risk of hepatocellular carcinoma in hepatitis B carriers. Gastroenterology 2003, 124, 327–334.
- Kao, J.H.; Chen, P.J.; Lai, M.Y.; Chen, D.S. Clinical and virological aspects of blood donors infected with hepatitis B virus genotypes B and C. J. Clin. Microbiol. 2002, 40, 22–25.
- Lin, C.L.; Liao, L.Y.; Liu, C.J.; Chen, P.J.; Lai, M.Y.; Kao, J.H.; Chen, D.S. Hepatitis B genotypes and precore/basal core promoter mutants in HBeAg-negative chronic hepatitis B. J. Gastroenterol. 2002, 37, 283–287.
- An, P.; Xu, J.; Yu, Y.; Winkler, C.A. Host and viral genetic variation in HBV-related hepatocellular carcinoma. Front. Genet. 2018, 9, 261.
- Wang, H.C.; Huang, W.; Lai, M.D.; Su, I.J. Hepatitis B virus pre-S mutants, endoplasmic reticulum stress and hepatocarcinogenesis. Cancer Sci. 2006, 97, 683–688.
- Choi, Y.M.; Lee, S.Y.; Kim, B.J. Naturally occurring hepatitis B virus mutations leading to endoplasmic reticulum stress and their contribution to the progression of hepatocellular carcinoma. Int. J. Mol. Sci. 2019, 20, 597.
- Yang, J.C.; Teng, C.F.; Wu, H.C.; Tsai, H.W.; Chuang, H.C.; Tsai, T.F.; Hsu, Y.H.; Huang, W.; Wu, L.W.; Su, I.J. Enhanced expression of vascular endothelial growth factor-A in ground glass hepatocytes and its implication in hepatitis B virus hepatocarcinogenesis. Hepatology 2009, 49, 1962–1971.
- Wang, H.C.; Chang, W.T.; Chang, W.W.; Wu, H.C.; Huang, W.; Lei, H.Y.; Lai, M.D.; Fausto, N.; Su, I.J. Hepatitis B virus pre-S2 mutant upregulates cyclin A expression and induces nodular proliferation of hepatocytes. Hepatology 2005, 41, 761–770.
- Luan, F.; Liu, H.; Gao, L.; Liu, J.; Sun, Z.; Ju, Y.; Hou, N.; Guo, C.; Liang, X.; Zhang, L.; et al. Hepatitis B virus protein preS2 potentially promotes HCC development via its transcriptional activation of hTERT. Gut 2009, 58, 1528–1537.
- Chisari, F.V.; Klopchin, K.; Moriyama, T.; Pasquinelli, C.; Dunsford, H.A.; Sell, S.; Pinkert, C.A.; Brinster, R.L.; Palmiter, R.D. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell 1989, 59, 1145–1156.
- Wang, L.H.; Huang, W.; Lai, M.D.; Su, I.J. Aberrant cyclin A expression and centrosome overduplication induced by hepatitis B virus pre-S2 mutants and its implication in hepatocarcinogenesis. Carcinogenesis 2012, 33, 466–472.
- Hung, J.H.; Su, I.J.; Lei, H.Y.; Wang, H.C.; Lin, W.C.; Chang, W.T.; Huang, W.; Chang, W.C.; Chang, Y.S.; Chen, C.C.; et al. Endoplasmic reticulum stress stimulates the expression of cyclooxygenase-2 through activation of NF-kappaB and pp38 mitogen-activated protein kinase. J. Biol. Chem. 2004, 279, 46384–46392.
- Hsieh, Y.H.; Su, I.J.; Wang, H.C.; Tsai, J.H.; Huang, Y.J.; Chang, W.W.; Lai, M.D.; Lei, H.Y.; Huang, W. Hepatitis B virus pre-S2 mutant surface antigen induces degradation of cyclin-dependent kinase inhibitor p27Kip1 through c-Jun activation domain-binding protein 1. Mol. Cancer Res. 2007, 5, 1063–1072.
- Tian, Y.; Sir, D.; Kuo, C.F.; Ann, D.K.; Ou, J.H. Autophagy required for hepatitis B virus replication in transgenic mice. J. Virol. 2011, 85, 13453–13456.
- Wu, B.K.; Li, C.C.; Chen, H.J.; Chang, J.L.; Jeng, K.S.; Chou, C.K.; Hsu, M.T.; Tsai, T.F. Blocking of G1/S transition and cell death in the regenerating liver of Hepatitis B virus X protein transgenic mice. Biochem. Biophys. Res. Commun. 2006, 340, 916–928.
- Kim, C.M.; Koike, K.; Saito, I.; Miyamura, T.; Jay, G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 1991, 351, 317–320.
- Yu, D.Y.; Moon, H.B.; Son, J.K.; Jeong, S.; Yu, S.L.; Yoon, H.; Han, Y.M.; Lee, C.S.; Park, J.S.; Lee, C.H.; et al. Incidence of hepatocellular carcinoma in transgenic mice expressing the hepatitis B virus X-protein. J. Hepatol. 1999, 31, 123–132.
- Zhu, H.; Wang, Y.; Chen, J.; Cheng, G.; Xue, J. Transgenic mice expressing hepatitis B virus X protein are more susceptible to carcinogen induced hepatocarcinogenesis. Exp. Mol. Pathol. 2004, 76, 44–50.
- Neuveut, C.; Wei, Y.; Buendia, M.A. Mechanisms of HBV-related hepatocarcinogenesis. J. Hepatol. 2010, 52, 594–604.
- Tang, H.; Delgermaa, L.; Huang, F.; Oishi, N.; Liu, L.; He, F.; Zhao, L.; Murakami, S. The transcriptional transactivation function of HBx protein is important for its augmentation role in hepatitis B virus replication. J. Virol. 2005, 79, 5548–5556.
- Keasler, V.V.; Hodgson, A.J.; Madden, C.R.; Slagle, B.L. Enhancement of hepatitis B virus replication by the regulatory X protein in vitro and in vivo. J. Virol. 2007, 81, 2656–2662.
- Decorsiere, A.; Mueller, H.; Van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 2016, 531, 386–389.
- Murphy, C.M.; Xu, Y.; Li, F.; Nio, K.; Reszka-Blanco, N.; Li, X.; Wu, Y.; Yu, Y.; Xiong, Y.; Su, L. Hepatitis B virus X protein promotes degradation of SMC5/6 to enhance HBV replication. Cell Rep. 2016, 16, 2846–2854.
- Potts, P.R.; Porteus, M.H.; Yu, H. Human SMC5/6 complex promotes sister chromatid homologous recombination by recruiting the SMC1/3 cohesin complex to double-strand breaks. EMBO J. 2006, 25, 3377–3388.
- Fujioka, Y.; Kimata, Y.; Nomaguchi, K.; Watanabe, K.; Kohno, K. Identification of a novel non-structural maintenance of chromosomes (SMC) component of the SMC5-SMC6 complex involved in DNA repair. J. Biol. Chem. 2002, 277, 21585–21591.
- Ampatzidou, E.; Irmisch, A.; O’Connell, M.J.; Murray, J.M. Smc5/6 is required for repair at collapsed replication forks. Mol. Cell. Biol. 2006, 26, 9387–9401.
- Piccoli, G.D.; Cortes-Ledesma, F.; Ira, G.; Torres-Rosell, J.; Uhle, S.; Farmer, S.; Hwang, J.Y.; Machin, F.; Ceschia, A.; McAleenan, A.; et al. Smc5-Smc6 mediate DNA double-strand-break repair by promoting sister-chromatid recombination. Nat. Cell Biol. 2006, 8, 1032–1034.
- Torres-Rosell, J.; Machin, F.; Farmer, S.; Jarmuz, A.; Eydmann, T.; Dalgaard, J.Z.; Aragon, L. SMC5 and SMC6 genes are required for the segregation of repetitive chromosome regions. Nat. Cell Biol. 2005, 7, 412–419.
- Livingston, C.M.; Ramakrishnan, D.; Strubin, M.; Fletcher, S.P.; Beran, R.K. Identifying and characterizing interplay between hepatitis B virus X protein and Smc5/6. Viruses 2017, 9, 69.
- Pereira, L.; Yi, F.; Merrill, B.J. Repression of nanog gene transcription by Tcf3 limits embryonic stem cell self-renewal. Mol. Cell. Biol. 2006, 26, 7479–7491.
- Takigawa, Y.; Brown, A.M. Wnt signaling in liver cancer. Curr. Drug Targets 2008, 9, 1013–1024.
- Polakis, P. The oncogenic activation of beta-catenin. Curr. Opin. Genet. Dev. 1999, 9, 15–21.
- Ding, Q.; Xia, W.; Liu, J.C.; Yang, J.Y.; Lee, D.F.; Xia, J.; Bartholomeusz, G.; Li, Y.; Pan, Y.; Li, Z.; et al. Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol. Cell 2005, 19, 159–170.
- Ho, T.C.; Wang, E.Y.; Yeh, K.H.; Jeng, Y.M.; Horng, J.H.; Wu, L.L.; Chen, Y.T.; Huang, H.C.; Hsu, C.L.; Chen, P.J.; et al. Complement C1q mediates the expansion of periportal hepatic progenitor cells in senescence-associated inflammatory liver. Proc. Natl. Acad. Sci. USA 2020, 117, 6717–6725.
- Wang, E.Y.; Yeh, S.H.; Tsai, T.F.; Huang, H.P.; Jeng, Y.M.; Lin, W.H.; Chen, W.C.; Yeh, K.H.; Chen, P.J.; Chen, D.S. Depletion of beta-catenin from mature hepatocytes of mice promotes expansion of hepatic progenitor cells and tumor development. Proc. Natl. Acad. Sci. USA 2011, 108, 18384–18389.
- Wang, K. Autophagy and apoptosis in liver injury. Cell Cycle 2015, 14, 1631–1642.
- Geng, M.; Xin, X.; Bi, L.Q.; Zhou, L.T.; Liu, X.H. Molecular mechanism of hepatitis B virus X protein function in hepatocarcinogenesis. World J. Gastroenterol. 2015, 21, 10732–10738.
- Lee, J.H.; Han, K.H.; Lee, J.M.; Park, J.H.; Kim, H.S. Impact of hepatitis B virus (HBV) x gene mutations on hepatocellular carcinoma development in chronic HBV infection. Clin. Vaccine Immunol. CVI 2011, 18, 914–921.
- Lin, C.L.; Chu, Y.D.; Yeh, C.T. Emergence of oncogenic-enhancing hepatitis B virus X gene mutants in patients receiving suboptimal entecavir treatment. Hepatology 2019, 69, 2292–2296.
- Riviere, L.; Quioc-Salomon, B.; Fallot, G.; Halgand, B.; Feray, C.; Buendia, M.A.; Neuveut, C. Hepatitis B virus replicating in hepatocellular carcinoma encodes HBx variants with preserved ability to antagonize restriction by Smc5/6. Antivir. Res. 2019, 172, 104618.
- Iavarone, M.; Trabut, J.B.; Delpuech, O.; Carnot, F.; Colombo, M.; Kremsdorf, D.; Brechot, C.; Thiers, V. Characterisation of hepatitis B virus X protein mutants in tumour and non-tumour liver cells using laser capture microdissection. J. Hepatol. 2003, 39, 253–261.
- Ma, N.F.; Lau, S.H.; Hu, L.; Xie, D.; Wu, J.; Yang, J.; Wang, Y.; Wu, M.C.; Fung, J.; Bai, X.; et al. COOH-terminal truncated HBV X protein plays key role in hepatocarcinogenesis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 5061–5068.
- Amaddeo, G.; Cao, Q.; Ladeiro, Y.; Imbeaud, S.; Nault, J.C.; Jaoui, D.; Mathe, Y.G.; Laurent, C.; Laurent, A.; Bioulac-Sage, P.; et al. Integration of tumour and viral genomic characterizations in HBV-related hepatocellular carcinomas. Gut 2015, 64, 820–829.
- Yu, M.W.; Chen, C.J. Hepatitis B and C viruses in the development of hepatocellular carcinoma. Crit. Rev. Oncol. Hematol. 1994, 17, 71–91.
- Yu, M.W.; Yang, Y.C.; Yang, S.Y.; Cheng, S.W.; Liaw, Y.F.; Lin, S.M.; Chen, C.J. Hormonal markers and hepatitis B virus-related hepatocellular carcinoma risk: A nested case-control study among men. J. Natl. Cancer Inst. 2001, 93, 1644–1651.
- Yu, M.W.; Cheng, S.W.; Lin, M.W.; Yang, S.Y.; Liaw, Y.F.; Chang, H.C.; Hsiao, T.J.; Lin, S.M.; Lee, S.D.; Chen, P.J.; et al. Androgen-receptor gene CAG repeats, plasma testosterone levels, and risk of hepatitis B-related hepatocellular carcinoma. J. Natl. Cancer Inst. 2000, 92, 2023–2028.
- Yu, M.W.; Chang, H.C.; Chang, S.C.; Liaw, Y.F.; Lin, S.M.; Liu, C.J.; Lee, S.D.; Lin, C.L.; Chen, P.J.; Lin, S.C.; et al. Role of reproductive factors in hepatocellular carcinoma: Impact on hepatitis B- and C-related risk. Hepatology 2003, 38, 1393–1400.
- Yang, W.J.; Chang, C.J.; Yeh, S.H.; Lin, W.H.; Wang, S.H.; Tsai, T.F.; Chen, D.S.; Chen, P.J. Hepatitis B virus X protein enhances the transcriptional activity of the androgen receptor through c-Src and glycogen synthase kinase-3β kinase pathways. Hepatology 2009, 49, 1515–1524.
- Chiu, C.M.; Yeh, S.H.; Chen, P.J.; Kuo, T.J.; Chang, C.J.; Chen, P.J.; Yang, W.J.; Chen, D.S. Hepatitis B virus X protein enhances androgen receptor-responsive gene expression depending on androgen level. Proc. Natl. Acad. Sci. USA 2007, 104, 2571–2578.
- Yu, Z.; Gao, Y.Q.; Feng, H.; Lee, Y.Y.; Li, M.S.; Tian, Y.; Go, M.Y.; Yu, D.Y.; Cheung, Y.S.; Lai, P.B.; et al. Cell cycle-related kinase mediates viral-host signalling to promote hepatitis B virus-associated hepatocarcinogenesis. Gut 2014, 63, 1793–1804.
- Wang, S.H.; Yeh, S.H.; Lin, W.H.; Wang, H.Y.; Chen, D.S.; Chen, P.J. Identification of androgen response elements in the enhancer I of hepatitis B virus: A mechanism for sex disparity in chronic hepatitis B. Hepatology 2009, 50, 1392–1402.
- Wang, S.H.; Yeh, S.H.; Chen, P.J. The driving circuit of HBx and androgen receptor in HBV-related hepatocarcinogenesis. Gut 2014, 63, 1688–1689.
- Chen, P.J.; Yeh, S.H.; Liu, W.H.; Lin, C.C.; Huang, H.C.; Chen, C.L.; Chen, D.S. Androgen pathway stimulates microRNA-216a transcription to suppress the tumor suppressor in lung cancer-1 gene in early hepatocarcinogenesis. Hepatology 2012, 56, 632–643.
- Feng, H.; Cheng, A.S.; Tsang, D.P.; Li, M.S.; Go, M.Y.; Cheung, Y.S.; Zhao, G.J.; Ng, S.S.; Lin, M.C.; Yu, J.; et al. Cell cycle-related kinase is a direct androgen receptor-regulated gene that drives beta-catenin/T cell factor-dependent hepatocarcinogenesis. J. Clin. Investig. 2011, 121, 3159–3175.
- Wang, S.H.; Yeh, S.H.; Shiau, C.W.; Chen, K.F.; Lin, W.H.; Tsai, T.F.; Teng, Y.C.; Chen, D.S.; Chen, P.J. Sorafenib action in hepatitis B virus X-activated oncogenic androgen pathway in liver through SHP-1. J. Natl. Cancer Inst. 2015, 107.
- Yan, Z.; Tan, W.; Xu, B.; Dan, Y.; Zhao, W.; Deng, C.; Chen, W.; Tan, S.; Mao, Q.; Wang, Y.; et al. A cis-acting regulatory variation of the estrogen receptor alpha (ESR1) gene is associated with hepatitis B virus-related liver cirrhosis. Hum. Mutat. 2011, 32, 1128–1136.
- Zhai, Y.; Zhou, G.; Deng, G.; Xie, W.; Dong, X.; Zhang, X.; Yu, L.; Yang, H.; Yuan, X.; Zhang, H.; et al. Estrogen receptor alpha polymorphisms associated with susceptibility to hepatocellular carcinoma in hepatitis B virus carriers. Gastroenterology 2006, 130, 2001–2009.
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007, 317, 121–124.
- Shimizu, I.; Kohno, N.; Tamaki, K.; Shono, M.; Huang, H.W.; He, J.H.; Yao, D.F. Female hepatology: Favorable role of estrogen in chronic liver disease with hepatitis B virus infection. World J. Gastroenterol. 2007, 13, 4295–4305.
- Sumi, D.; Hayashi, T.; Matsui-Hirai, H.; Jacobs, A.T.; Ignarro, L.J.; Iguchi, A. 17beta-estradiol inhibits NADPH oxidase activity through the regulation of p47phox mRNA and protein expression in THP-1 cells. Biochim. Biophys. Acta 2003, 1640, 113–118.
- Wen, Y.; Yang, S.; Liu, R.; Perez, E.; Yi, K.D.; Koulen, P.; Simpkins, J.W. Estrogen attenuates nuclear factor-kappa B activation induced by transient cerebral ischemia. Brain Res. 2004, 1008, 147–154.
- Wang, S.H.; Yeh, S.H.; Lin, W.H.; Yeh, K.H.; Yuan, Q.; Xia, N.S.; Chen, D.S.; Chen, P.J. Estrogen receptor α represses transcription of HBV genes via interaction with hepatocyte nuclear factor 4α. Gastroenterology 2012, 142, 989–998.e4.
- Wang, S.H.; Chen, P.J.; Yeh, S.H. Gender disparity in chronic hepatitis B: Mechanisms of sex hormones. J. Gastroenterol. Hepatol. 2015, 23, 63–69.
- Liu, W.H.; Yeh, S.H.; Lu, C.C.; Yu, S.L.; Chen, H.Y.; Lin, C.Y.; Chen, D.S.; Chen, P.J. MicroRNA-18a prevents estrogen receptor-alpha expression, promoting proliferation of hepatocellular carcinoma cells. Gastroenterology 2009, 136, 683–693.
- Tsukuda, S.; Watashi, K. Hepatitis B virus biology and life cycle. Antivir. Res. 2020, 182, 104925.
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA integration: Molecular mechanisms and clinical implications. Viruses 2017, 9, 75.
- Mason, W.S.; Low, H.C.; Xu, C.; Aldrich, C.E.; Scougall, C.A.; Grosse, A.; Clouston, A.; Chavez, D.; Litwin, S.; Peri, S.; et al. Detection of clonally expanded hepatocytes in chimpanzees with chronic hepatitis B virus infection. J. Virol. 2009, 83, 8396–8408.
- Tokino, T.; Matsubara, K. Chromosomal sites for hepatitis B virus integration in human hepatocellular carcinoma. J. Virol. 1991, 65, 6761–6764.
- Matsubara, K.; Tokino, T. Integration of hepatitis B virus DNA and its implications for hepatocarcinogenesis. Mol. Biol. Med. 1990, 7, 243–260.
- Li, C.L.; Li, C.Y.; Lin, Y.Y.; Ho, M.C.; Chen, D.S.; Chen, P.J.; Yeh, S.H. Androgen receptor enhances hepatic telomerase reverse transcriptase gene transcription after hepatitis B virus integration or point mutation in promoter region. Hepatology 2019, 69, 498–512.
- Jiang, Z.; Jhunjhunwala, S.; Liu, J.; Haverty, P.M.; Kennemer, M.I.; Guan, Y.; Lee, W.; Carnevali, P.; Stinson, J.; Johnson, S.; et al. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res. 2012, 22, 593–601.
- Guerrero, R.B.; Roberts, L.R. The role of hepatitis B virus integrations in the pathogenesis of human hepatocellular carcinoma. J. Hepatol. 2005, 42, 760–777.
- Hessein, M.; Saad, E.G.; Mohamed, A.A.; Kamel, E.A.M.; Hady, A.M.A.; Amina, M.; Rogler, C.E. Hit-and-run mechanism of HBV-mediated progression to hepatocellular carcinoma. Tumori 2005, 91, 241–247.
- Sherr, C.J. Principles of tumor suppression. Cell 2004, 116, 235–246.
- Bedogni, G.; Miglioli, L.; Masutti, F.; Ferri, S.; Castiglione, A.; Lenzi, M.; Croce, L.S.; Granito, A.; Tiribelli, C.; Bellentani, S. Natural course of chronic HCV and HBV infection and role of alcohol in the general population: The Dionysos Study. Am. J. Gastroenterol. 2008, 103, 2248–2253.
- Iida-Ueno, A.; Enomoto, M.; Tamori, A.; Kawada, N. Hepatitis B virus infection and alcohol consumption. World J. Gastroenterol. 2017, 23, 2651–2659.
- Li, Z.M.; Kong, C.Y.; Zhang, S.L.; Han, B.; Zhang, Z.Y.; Wang, L.S. Alcohol and HBV synergistically promote hepatic steatosis. Ann. Hepatol. 2019, 18, 913–917.
- Ganesan, M.; Eikenberry, A.; Poluektova, L.Y.; Kharbanda, K.K.; Osna, N.A. Role of alcohol in pathogenesis of hepatitis B virus infection. World J. Gastroenterol. 2020, 26, 883–903.

