1. Introduction
Hepatocellular carcinoma (HCC), accounting for 90% of primary liver cancer cases, is one of the common malignancies worldwide since the incidence ranks sixth, and the mortality rate ranks third among all cancers. Due to the lack of specific disease symptoms and reliable diagnostic markers at early stages, HCC is still considered a challenging public health issue. Most HCC cases are usually diagnosed in an advanced stage, and this generally restricts the efficacy of therapies. Compared with other gastrointestinal tract tumors, the prognosis of HCC patients is relatively poor since the 5-year survival rate is less than 20%
[1]. Among all etiologic factors attributing to HCC development, chronic hepatitis B virus (HBV) infection is the most important risk factor, which accounts for around 50% of HCC cases overall
[2].
The discovery of HBV around 50 years ago leads an intensive investigation of HBV virology, immunology, and pathogenesis, which also lay down the base for the development of an effective vaccine. The implementation of HBV vaccination reduces 90% of chronic hepatitis B (CHB) prevalence in the vaccinated cohorts, as shown in Taiwan and other Asia countries
[3][4][5][3,4,5], which results in a parallel decline of young-age HCC
[6][7][6,7]. Despite the universal vaccination program being implemented for 35 years, there are still about 257 million CHB carriers by WHO estimation
[8].
HBV infection stimulates hepatocarcinogenesis via multiple routes, especially through inducing persistent chronic inflammation
[9]. About 15% to 40% of CHB patients eventually proceed into end-stage liver diseases, including cirrhosis and HCC
[10]. Clinical use of antiviral nucleotide and nucleot(s)tide analogs (NUCs) at the first line efficiently represses HBV replication and reduces inflammation
[11][12][11,12]. Subsequently, the HCC risk in these NUCs-treated patients is significantly lowered
[13][14][13,14]. Therefore, long-term antiviral therapies by NUCs become the standard of care for CHB. However, the residual HCC risk in these treated patients is not negligible since the five-year cumulative incidence of HCC in the noncirrhotic population remains up to 6.9%, which is still above the threshold of surveillance
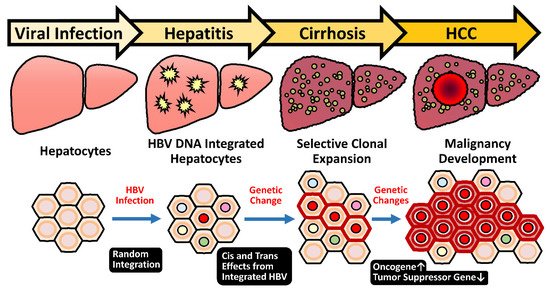
[14][15][14,15]. One of the reasons is probably due to the unique feature of HBV DNA integration that happens during early infection periods in the majority of HBV-related HCC
[16]. The event of HBV integration is not a mandatory process during the HBV life cycle that proceeds through the episomal, covalently linked close-circular DNA (cccDNA). Therefore, the incidental HBV integration into host chromosomes occurs in only 0.1% of infected hepatocytes
[17][18][17,18], but it is present in 90% of HBV-related HCC
[19]. The dominance of HBV integration in the HBV-related HCC strongly implicates its carcinogenic potential.
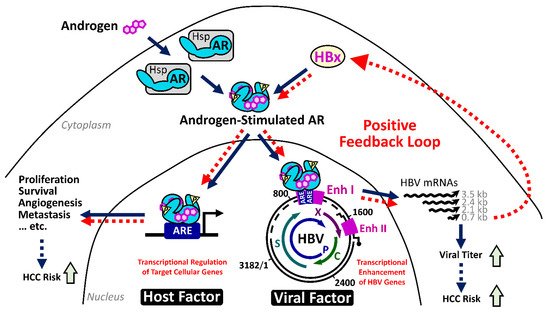
Since only about a quarter of CHB patients will succumb to HCC, numerous studies have investigated the risk factors from virus and host for HCC development. The viral risk factors include higher viral titer, HBeAg-positive status, specific viral genotypes, and genetic mutations in viral genomes
[9]. The host factors contain older age, male gender, familial history, and genetic variants of immune response genes
[20]. Among these risk factors, the male gender has been noted as a striking feature in HBV-related HCC. Slight male gender preference is noted in nonviral HCC patients, which, however, is more evident for the HCC cases in chronic hepatitis B (CHB) pandemic areas
[21]. The male-to-female ratio ranges from 5:1 to 7:1 for HBV-related HCC but only around 2.8:1 for hepatitis C virus-related HCC
[22]. It is noteworthy that this gender difference in the carcinogenic process is progressively increasing during disease evolution. An epidemiological study in Taiwan pointed out that in the asymptomatic carriers, the male-to-female ratio was about 1.2:1, then was increased to 6.3:1 in patients at CHB stage, and finally became 9.8:1 in HCC patients
[23]. In a large cohort community-based case–control study in Taiwan, the male CHB patients carried higher HBV viral loads than their female counterparts
[24]. Even in the vaccinated cohort, which has been followed up for over 18 years, the prevalence of occult HBV infection was higher in males than in females (10.7% vs. 4.4%)
[25]. Therefore, the male gender is believed to be a major risk factor for HBV-related hepatocarcinogenesis, starting from the relatively early chronic hepatitis stage and retained until the late stage of HCC development.
This review article focuses on the unique HBV-induced hepatocarcinogenic mechanisms in detail, covering both male gender and HBV integration, which may provide new insights for medical development in the related fields of pathogenesis, diagnosis, and treatment.