 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Manoj Kumar Jena | + 6911 word(s) | 6911 | 2021-06-10 08:51:45 | | | |
| 2 | Vivi Li | Meta information modification | 6911 | 2021-06-11 09:51:22 | | |
Video Upload Options
Preeclampsia (PE) is a serious pregnancy complication, affecting about 5–7% of pregnancies worldwide and is characterized by hypertension and damage to multiple maternal organs, primarily the liver and kidneys. PE usually begins after 20 weeks’ gestation and, if left untreated, can lead to serious complications and lifelong disabilities—even death—in both the mother and the infant. As delivery is the only cure for the disease, treatment is primarily focused on the management of blood pressure and other clinical symptoms. The pathogenesis of PE is still not clear. Abnormal spiral artery remodeling, placental ischemia and a resulting increase in the circulating levels of vascular endothelial growth factor receptor-1 (VEGFR-1), also called soluble fms-like tyrosine kinase-1 (sFlt-1), are believed to be among the primary pathologies associated with PE. sFlt-1 is produced mainly in the placenta during pregnancy and acts as a decoy receptor, binding to free VEGF (VEGF-A) and placental growth factor (PlGF), resulting in the decreased bioavailability of each to target cells. Despite the pathogenic effects of increased sFlt-1 on the maternal vasculature, recent studies from our laboratory and others have strongly indicated that the increase in sFlt-1 in PE may fulfill critical protective functions in preeclamptic pregnancies.
1. Introduction
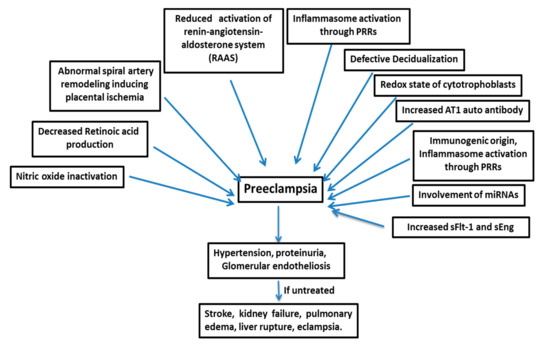
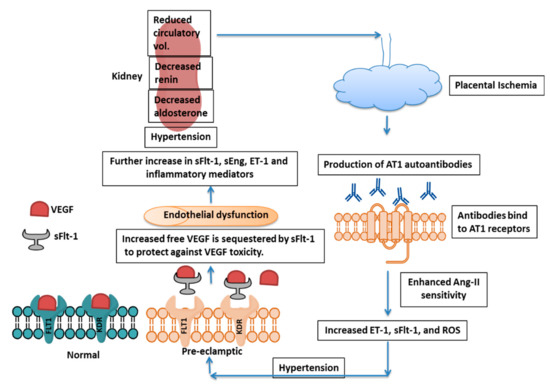
2. Pathogenesis of Preeclampsia
2.1. sFlt-1: The Central Molecule in PE
2.2. Factors Regulating sFlt-1 Expression
2.3. Impeded Spiral Artery Remodeling
2.4. Redox State of Cytotrophoblasts
2.5. Angiotensin-II Type 1 Receptor Autoantibody
2.6. Inflammatory Cytokines in PE Pathogenesis
2.7. Signaling Pathways Involved
2.8. The Role of miRNAs and lncRNAs in Pathogenesis
2.9. The Role of Endothelin-1 (ET-1) in the Pathophysiology of PE
2.10. Miscellaneous Factors
3. Therapeutic Approaches
References
- Rana, S.; Lemoine, E.; Granger, J.P.; Karumanchi, A. Preeclampsia: Pathophysiology, Challenges, and Perspectives. Circ. Res. 2019, 124, 1094–1112.
- Hogan, M.; Foreman, K.; Naghavi, M.; Ahn, S.; Wang, M.; Makela, S.; Lopez, A.; Lozano, R.; Murray, C.J. Maternal Mortality for 181 Countries, 1980–2008. Obstet. Anesthesia Dig. 2011, 31, 69.
- Wanderer, J.P.; Leffert, L.R.; Mhyre, J.M.; Kuklina, E.V.; Callaghan, W.M.; Bateman, B.T. Epidemiology of Obstetric-Related ICU Admissions in Maryland. Crit. Care Med. 2013, 41, 1844–1852.
- Kuklina, E.V.; Ayala, C.; Callaghan, W.M. Hypertensive Disorders and Severe Obstetric Morbidity in the United States. Obstet. Gynecol. 2009, 113, 1299–1306.
- Coutinho, T.; Lamai, O.; Nerenberg, K. Hypertensive Disorders of Pregnancy and Cardiovascular Diseases: Current Knowledge and Future Directions. Curr. Treat. Options Cardiovasc. Med. 2018, 20, 56.
- Tooher, J.; Thornton, C.; Makris, A.; Ogle, R.; Korda, A.; Hennessy, A. All Hypertensive Disorders of Pregnancy Increase the Risk of Future Cardiovascular Disease. Hypertension 2017, 70, 798–803.
- Ilekis, J.V.; Reddy, U.M.; Roberts, J.M. Review Article: Preeclampsia—A Pressing Problem: An Executive Summary of a National Institute of Child Health and Human Development Workshop. Reprod. Sci. 2007, 14, 508–523.
- Redman, C.; Sargent, I.; Staff, A. IFPA Senior Award Lecture: Making sense of pre-eclampsia—Two placental causes of preeclampsia? Placenta 2014, 35, S20–S25.
- Young, B.C.; Levine, R.J.; Karumanchi, S.A. Pathogenesis of Preeclampsia. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 173–192.
- Cormick, G.; Betran, A.P.; Ciapponi, A.; Hall, D.R.; Hofmeyr, J. Calcium and Pre-eclampsia Study Group; on behalf of the Calcium and Pre-eclampsia Study Group Inter-pregnancy interval and risk of recurrent pre-eclampsia: Systematic review and meta-analysis. Reprod. Heal. 2016, 13, 83.
- Maynard, S.E.; Min, J.Y.; Merchan, J.; Lim, K.H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Investig. 2003, 111, 649–658.
- Leavey, K.; Bainbridge, S.A.; Cox, B.J. Large Scale Aggregate Microarray Analysis Reveals Three Distinct Molecular Subclasses of Human Preeclampsia. PLoS ONE 2015, 10, e0116508.
- Leavey, K.; Benton, S.J.; Grynspan, D.; Kingdom, J.C.; Bainbridge, S.A.; Cox, B.J. Unsupervised Placental Gene Expression Profiling Identifies Clinically Relevant Subclasses of Human Preeclampsia. Hypertension 2016, 68, 137–147.
- Than, N.G.; Romero, R.; Tarca, A.L.; Kekesi, K.A.; Xu, Y.; Xu, Z.; Juhasz, K.; Bhatti, G.; Leavitt, R.J.; Gelencser, Z.; et al. Integrated Systems Biology Approach Identifies Novel Maternal and Placental Pathways of Preeclampsia. Front. Immunol. 2018, 9, 1661.
- Bartsch, E.; Medcalf, K.E.; Park, A.L.; Ray, J.G. Clinical risk factors for pre-eclampsia determined in early pregnancy: Systematic review and meta-analysis of large cohort studies. BMJ 2016, 353, 1753.
- Sahu, M.B.; Deepak, V.; Gonzales, S.K.; Rimawi, B.H.; Watkins, K.K.; Smith, A.K.; Badell, M.L.; Sidell, N.; Rajakumar, A. Decidual cells from women with preeclampsia exhibit inadequate decidualization and reduced sFlt1 suppression. Pregnancy Hypertens. 2018, 15, 64–71.
- D’Souza, V.; Rani, A.; Patil, V.; Pisal, H.; Randhir, K.; Mehendale, S.; Wagh, G.; Gupte, S.; Joshi, S. Increased oxidative stress from early pregnancy in women who develop preeclampsia. Clin. Exp. Hypertens. 2016, 38, 225–232.
- Nevo, O.; Soleymanlou, N.; Wu, Y.; Xu, J.; Kingdom, J.; Many, A.; Zamudio, S.; Caniggia, I. Increased expression of sFlt-1 in in vivo and in vitro models of human placental hypoxia is mediated by HIF-1. Am. J. Physiol. Integr. Comp. Physiol. 2006, 291, R1085–R1093.
- George, E.M.; Cockrell, K.; Adair, T.H.; Granger, J. Regulation of sFlt-1 and VEGF secretion by adenosine under hypoxic conditions in rat placental villous explants. Am. J. Physiol. Integr. Comp. Physiol. 2010, 299, R1629–R1633.
- Hunter, A.; Aitkenhead, M.; Caldwell, C.; McCracken, G.; Wilson, D.; McClure, N. Serum levels of vascular endothelial growth factor in preeclamptic and normotensive pregnancy. Hypertension 2000, 36, 965–969.
- Neufeld, G.; Cohen, T.; Gengrinovitch, S.; Poltorak, Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999, 13, 9–22.
- Shibuya, M. Structure and function of VEGF/VEGF-receptor system involved in angiogenesis. Cell Struct. Funct. 2001, 26, 25–35.
- Kliche, S.; Waltenberger, J. VEGF Receptor Signaling and Endothelial Function. IUBMB Life 2001, 52, 61–66.
- Luttun, A.; Tjwa, M.; Moons, L.; Wu, Y.; Angelillo-Scherrer, A.; Liao, F.; Nagy, J.A.; Hooper, A.; Priller, J.; De Klerck, B.; et al. Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat. Med. 2002, 8, 831–840.
- Saito, T.; Takeda, N.; Amiya, E.; Nakao, T.; Abe, H.; Semba, H.; Soma, K.; Koyama, K.; Hosoya, Y.; Imai, Y.; et al. VEGF-A induces its negative regulator, soluble form of VEGFR-1, by modulating its alternative splicing. FEBS Lett. 2013, 587, 2179–2185.
- Seki, H. Balance of antiangiogenic and angiogenic factors in the context of the etiology of preeclampsia. Acta Obstet. Gynecol. Scand. 2014, 93, 959–964.
- Kendall, R.L.; Wang, G.; Thomas, K.A. Identification of a Natural Soluble Form of the Vascular Endothelial Growth Factor Receptor, FLT-1, and Its Heterodimerization with KDR. Biochem. Biophys. Res. Commun. 1996, 226, 324–328.
- Kendall, R.L.; Thomas, K.A. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 10705–10709.
- Vogtmann, R.; Kühnel, E.; Dicke, N.; Verkaik-Schakel, R.N.; Plösch, T.; Schorle, H.; Stojanovska, V.; Herse, F.; Köninger, A.; Kimmig, R.; et al. Human sFLT1 leads to Severe Changes in Placental Differentiation and Vascularization in a Transgenic hsFLT1/rtTA FGR Mouse Model. Front. Endocrinol. 2019, 10, 165.
- Mustonen, T.; Alitalo, K. Endothelial receptor tyrosine kinases involved in angiogenesis. J. Cell Biol. 1995, 129, 895–898.
- Roberts, J.M.; Rajakumar, A. Preeclampsia and soluble fms-like tyrosine kinase 1. J. Clin. Endocrinol. Metab. 2009, 94, 2252–2254.
- Zhou, Y.; McMaster, M.; Woo, K.; Janatpour, M.; Perry, J.; Karpanen, T.; Alitalo, K.; Damsky, C.; Fisher, S.J. Vascular Endothelial Growth Factor Ligands and Receptors That Regulate Human Cytotrophoblast Survival Are Dysregulated in Severe Preeclampsia and Hemolysis, Elevated Liver Enzymes, and Low Platelets Syndrome. Am. J. Pathol. 2002, 160, 1405–1423.
- Thadhani, R.; Kisner, T.; Hagmann, H.; Bossung, V.; Noack, S.; Schaarschmidt, W.; Jank, A.; Kribs, A.; Cornely, O.A.; Kreyssig, C.; et al. Pilot Study of Extracorporeal Removal of Soluble Fms-Like Tyrosine Kinase 1 in Preeclampsia. Circulation 2011, 124, 940–950.
- Clark, D.E.; Smith, S.; He, Y.; Day, K.A.; Licence, D.R.; Corps, A.N.; Lammoglia, R.; Charnock-Jones, D.S. A vascular endothelial growth factor antagonist is produced by the human placenta and released into the maternal circulation. Biol. Reprod. 1998, 59, 1540–1548.
- Levine, R.J.; Maynard, S.E.; Qian, C.; Lim, K.-H.; England, L.J.; Yu, K.F.; Schisterman, E.F.; Thadhani, R.; Sachs, B.P.; Epstein, F.H.; et al. Circulating Angiogenic Factors and the Risk of Preeclampsia. N. Engl. J. Med. 2004, 350, 672–683.
- McMahon, K.; Karumanchi, S.A.; Stillman, I.E.; Cummings, P.; Patton, D.; Easterling, T. Does soluble fms-like tyrosine kinase-1 regulate placental invasion? Insight from the invasive placenta. Am. J. Obstet. Gynecol. 2014, 210, 68.e1–68.e4.
- Hirashima, M.; Lu, Y.; Byers, L.; Rossant, J. Trophoblast expression of fms-like tyrosine kinase 1 is not required for the establishment of the maternal–fetal interface in the mouse placenta. Proc. Natl. Acad. Sci. USA 2003, 100, 15637–15642.
- Palmer, K.R.; Tong, S.; Tuohey, L.; Cannon, P.; Ye, L.; Hannan, N.J.; Brownfoot, F.C.; Illanes, S.E.; Kaitu’U-Lino, T.J. Jumonji Domain Containing Protein 6 Is Decreased in Human Preeclamptic Placentas and Regulates sFLT-1 Splice Variant Production1. Biol. Reprod. 2016, 94, 59.
- Boeckel, J.-N.; Guarani, V.; Koyanagi, M.; Roexe, T.; Lengeling, A.; Schermuly, R.T.; Gellert, P.; Braun, T.; Zeiher, A.; Dimmeler, S. Jumonji domain-containing protein 6 (Jmjd6) is required for angiogenic sprouting and regulates splicing of VEGF-receptor 1. Proc. Natl. Acad. Sci. USA 2011, 108, 3276–3281.
- Loenarz, C.; Schofield, C.J. Expanding chemical biology of 2-oxoglutarate oxygenases. Nat. Methods 2008, 4, 152–156.
- Pollard, P.; Loenarz, C.; Mole, D.R.; McDonough, M.A.; Gleadle, J.; Schofield, C.J.; Ratcliffe, P.J. Regulation of Jumonji-domain-containing histone demethylases by hypoxia-inducible factor (HIF)-1α. Biochem. J. 2008, 416, 387–394.
- Tsukada, Y.-I.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2005, 439, 811–816.
- Palmer, K.R.; Tong, S.; Kaitu’U-Lino, T.J. Placental-specific sFLT-1: Role in pre-eclamptic pathophysiology and its translational possibilities for clinical prediction and diagnosis. Mol. Hum. Reprod. 2016, 23, 69–78.
- Palmer, K.R.; Kaitu’U-Lino, T.J.; Hastie, R.; Hannan, N.J.; Ye, L.; Binder, N.; Cannon, P.; Tuohey, L.; Johns, T.; Shub, A.; et al. Placental-Specific sFLT-1 e15a Protein Is Increased in Preeclampsia, Antagonizes Vascular Endothelial Growth Factor Signaling, and Has Antiangiogenic Activity. Hypertension 2015, 66, 1251–1259.
- Szalai, G.; Xu, Y.; Romero, R.; Chaiworapongsa, T.; Xu, Z.; Chiang, P.J.; Ahn, H.; Sundell, B.; Plazyo, O.; Jiang, Y.; et al. In Vivo Experiments Reveal the Good, the Bad and the Ugly Faces of sFlt-1 in Pregnancy. PLoS ONE 2014, 9, e110867.
- Bergmann, A.; Ahmad, S.; Cudmore, M.; Gruber, A.D.; Wittschen, P.; Lindenmaier, W.; Christofori, G.; Gross, V.; Gonzalves, A.C.D.C.; Gröne, H.-J.; et al. Reduction of circulating soluble Flt-1 alleviates preeclampsia-like symptoms in a mouse model. J. Cell. Mol. Med. 2009, 14, 1857–1867.
- Ahmad, S.; Ahmed, A. Elevated Placental Soluble Vascular Endothelial Growth Factor Receptor-1 Inhibits Angiogenesis in Preeclampsia. Circ. Res. 2004, 95, 884–891.
- Powers, R.; Roberts, J.; Cooper, K.; Gallaher, M.; Frank, M.; Harger, G.; Ness, R. Maternal serum soluble fms-like tyrosine kinase 1 concentrations are not increased in early pregnancy and decrease more slowly postpartum in women who develop preeclampsia. Am. J. Obstet. Gynecol. 2005, 193, 185–191.
- Jiang, Z.; Zou, Y.; Ge, Z.; Zuo, Q.; Huang, S.Y.; Sun, L. A Role of sFlt-1 in Oxidative Stress and Apoptosis in Human and Mouse Pre-Eclamptic Trophoblasts1. Biol. Reprod. 2015, 93, 73.
- Cohen, J.M.; Kramer, M.S.; Platt, R.W.; Basso, O.; Evans, R.W.; Kahn, S.R. The association between maternal antioxidant levels in midpregnancy and preeclampsia. Am. J. Obstet. Gynecol. 2015, 213, 695.e1–695.e13.
- Miquerol, L.; Langille, B.L.; Nagy, A. Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development 2000, 127, 3941–3946.
- Fan, X.; Rai, A.; Kambham, N.; Sung, J.F.; Singh, N.; Petitt, M.; Dhal, S.; Agrawal, R.; Sutton, R.E.; Druzin, M.L.; et al. Endometrial VEGF induces placental sFLT1 and leads to pregnancy complications. J. Clin. Investig. 2014, 124, 4941–4952.
- Murakami, Y.; Kobayashi, T.; Omatsu, K.; Suzuki, M.; Ohashi, R.; Matsuura, T.; Sugimura, M.; Kanayama, N. Exogenous Vascular Endothelial Growth Factor Can Induce Preeclampsia-Like Symptoms in Pregnant Mice. Semin. Thromb. Hemost. 2005, 31, 307–313.
- Parchem, J.G.; Kanasaki, K.; Kanasaki, M.; Sugimoto, H.; Xie, L.; Hamano, Y.; Lee, S.B.; Gattone, V.H.; Parry, S.; Strauss, J.F.; et al. Loss of placental growth factor ameliorates maternal hypertension and preeclampsia in mice. J. Clin. Investig. 2018, 128, 5008–5017.
- Chng, S.C.; Ho, L.; Tian, J.; Reversade, B. ELABELA: A Hormone Essential for Heart Development Signals via the Apelin Receptor. Dev. Cell 2013, 27, 672–680.
- Ho, L.; Tan, S.Y.X.; Wee, S.; Wu, Y.; Tan, S.J.C.; Ramakrishna, N.B.; Chng, S.C.; Nama, S.; Szczerbinska, I.; Chan, Y.S.; et al. ELABELA is an endogenous growth factor that sustains hESC self-renewal via the PI3K/AKT pathway. Cell Stem Cell 2015, 17, 435–447.
- Bertrand, C.; Valet, P.; Castan-Laurell, I. Apelin and energy metabolism. Front. Physiol. 2015, 6, 115.
- Yamaleyeva, L.M.; Shaltout, H.A.; Varagic, J. Apelin-13 in blood pressure regulation and cardiovascular disease. Curr. Opin. Nephrol. Hypertens. 2016, 25, 396–403.
- Mughal, A.; O’Rourke, S.T. Vascular effects of apelin: Mechanisms and therapeutic potential. Pharmacol. Ther. 2018, 190, 139–147.
- Wang, Z.; Yu, D.; Wang, M.; Wang, Q.; Kouznetsova, J.; Yang, R.; Qian, K.; Wu, W.; Shuldiner, A.R.; Sztalryd, C.; et al. Elabela-Apelin Receptor Signaling Pathway is Functional in Mammalian Systems. Sci. Rep. 2015, 5, 8170.
- Georgiadou, D.; Afink, G.B.; Van Dijk, M. The apelinergic-axis in human preeclamptic pregnancies: A systematic review. Pregnancy Hypertens. 2019, 17, 148–157.
- Eberlé, D.; Marousez, L.; Hanssens, S.; Knauf, C.; Breton, C.; Deruelle, P.; Lesage, J. Elabela and Apelin actions in healthy and pathological pregnancies. Cytokine Growth Factor Rev. 2019, 46, 45–53.
- Zhou, L.; Sun, H.; Cheng, R.; Fan, X.; Lai, S.; Deng, C. ELABELA, as a potential diagnostic biomarker of preeclampsia, regulates abnormally shallow placentation via APJ. Am. J. Physiol. Metab. 2019, 316, E773–E781.
- Xiao, Z.; Li, S.; Yu, Y.; Li, M.; Chen, J.; Wang, F.; Zhang, J.; Deng, W.; Yang, Q.; Fan, X. VEGF-A regulates sFlt-1 production in trophoblasts through both Flt-1 and KDR receptors. Mol. Cell. Biochem. 2018, 449, 1–8.
- Deepak, V.; Sahu, M.B.; Yu, J.; Jones, J.W.; Kane, M.A.; Taylor, R.N.; Badell, M.L.; Sidell, N.; Rajakumar, A. Retinoic Acid Is a Negative Regulator of sFLT1 Expression in Decidual Stromal Cells, and Its Levels Are Reduced in Preeclamptic Decidua. Hypertension 2019, 73, 1104–1111.
- Gonçalves-Rizzi, V.H.; Possomato-Vieira, J.S.; Graça, T.U.S.; Nascimento, R.A.; Dias-Junior, C.A. Sodium nitrite attenuates hypertension-in-pregnancy and blunts increases in soluble fms-like tyrosine kinase-1 and in vascular endothelial growth factor. Nitric Oxide 2016, 57, 71–78.
- Shashar, M.; Zubkov, A.; Chernichovski, T.; Hershkovitz, R.; Hoffman, E.; Grupper, A.; Weinstein, T.; Schwartz, I.F. Profound Decrease in Glomerular Arginine Transport by CAT (Cationic Amino Acid Transporter)-1 Contributes to the FLT-1 (FMS-Like Tyrosine Kinase 1) Induced Preeclampsia in the Pregnant Mice. Hypertension 2019, 73, 878–884.
- Liu, S.; Premont, R.T.; Rockey, D.C. G-protein-coupled Receptor Kinase Interactor-1 (GIT1) Is a New Endothelial Nitric-oxide Synthase (eNOS) Interactor with Functional Effects on Vascular Homeostasis. J. Biol. Chem. 2012, 287, 12309–12320.
- Zhang, S.; Zou, C.; Zhang, Q. Deletion of GIT1 Impacts eNOS Activity To Aggravate sFlt-1–Induced Preeclampsia Phenotype in Mice. G3 Genes|Genomes|Genetics 2018, 8, 3377–3382.
- Eddy, A.C.; Chapman, H.; George, E.M. Heparanase regulation of sFLT-1 release in trophoblasts in vitro. Placenta 2019, 85, 63–68.
- Cottrell, H.N.; Wu, J.; Rimawi, B.H.; Duran, J.M.; Spencer, J.B.; Sidell, N.; Rajakumar, A. Human endometrial stromal cell plasticity: Reversible sFlt1 expression negatively coincides with decidualization. Hypertens. Pregnancy 2017, 78, 1–8.
- Liu, J.; Ji, X.; Li, Z.; Yang, X.; Wang, W.; Zhang, X. G protein gamma subunit 7 induces autophagy and inhibits cell division. Oncotarget 2016, 7, 24832.
- Gilbert, J.S.; Ryan, M.J.; Lamarca, B.B.; Sedeek, M.; Murphy, S.R.; Granger, J.P. Pathophysiology of hypertension during preeclampsia: Linking placental ischemia with endothelial dysfunction. Am. J. Physiol. Circ. Physiol. 2008, 294, H541–H550.
- Granger, J.P.; Alexander, B.T.; Llinas, M.T.; Bennett, W.A.; Khalil, R.A. Pathophysiology of preeclampsia: Linking placental ischemia/hypoxia with microvascular dysfunction. Microcirculation 2002, 9, 147–160.
- Murphy, S.R.; Lamarca, B.B.D.; Parrish, M.; Cockrell, K.; Granger, J. Control of soluble fms-like tyrosine-1 (sFlt-1) production response to placental ischemia/hypoxia: Role of tumor necrosis factor-α. Am. J. Physiol. Integr. Comp. Physiol. 2012, 304, R130–R135.
- Vaka, V.R.; McMaster, K.M.; Cunningham, M.W.; Ibrahim, T.; Hazlewood, R.; Usry, N.; Cornelius, D.C.; Amaral, L.M.; Lamarca, B.D. Role of Mitochondrial Dysfunction and Reactive Oxygen Species in Mediating Hypertension in the Reduced Uterine Perfusion Pressure Rat Model of Preeclampsia. Hypertension 2018, 72, 703–711.
- Covarrubias, A.; LeCarpentier, E.; Lo, A.; Salahuddin, S.; Gray, K.J.; Karumanchi, S.A.; Zsengellér, Z.K. AP39, a Modulator of Mitochondrial Bioenergetics, Reduces Antiangiogenic Response and Oxidative Stress in Hypoxia-Exposed Trophoblasts. Am. J. Pathol. 2019, 189, 104–114.
- Tang, B.; Guller, S.; Gurpide, E. Mechanisms involved in the decidualization of human endometrial stromal cells. Acta Eur. Fertil. 1993, 24, 221–223.
- Dunn, C.L.; Kelly, R.W.; Critchley, H.O.D. Decidualization of the human endometrial stromal cell: An enigmatic transformation. Reprod. Biomed. Online 2003, 7, 151–161.
- Schatz, F.; Guzeloglu-Kayisli, O.; Arlier, S.; Kayisli, U.A.; Lockwood, C.J. The role of decidual cells in uterine hemostasis, menstruation, inflammation, adverse pregnancy outcomes and abnormal uterine bleeding. Hum. Reprod. Updat. 2016, 22, 497–515.
- Lamarca, B.D.; Alexander, B.T.; Gilbert, J.S.; Ryan, M.J.; Sedeek, M.; Murphy, S.R.; Granger, J.P. Pathophysiology of hypertension in response to placental ischemia during pregnancy: A central role for endothelin? Gend. Med. 2008, 5, S133–S138.
- Verdonk, K.; Saleh, L.; Lankhorst, S.; Smilde, J.I.; Van Ingen, M.M.; Garrelds, I.M.; Friesema, E.C.; Russcher, H.; Meiracker, A.H.V.D.; Visser, W.; et al. Association Studies Suggest a Key Role for Endothelin-1 in the Pathogenesis of Preeclampsia and the Accompanying Renin–Angiotensin–Aldosterone System Suppression. Hypertension 2015, 65, 1316–1323.
- Zhao, Y.; Koga, K.; Osuga, Y.; Nagai, M.; Izumi, G.; Takamura, M.; Harada, M.; Hirota, Y.; Yoshino, O.; Taketani, Y. Thrombin enhances soluble Fms-like tyrosine kinase 1 expression in trophoblasts; possible involvement in the pathogenesis of preeclampsia. Fertil. Steril. 2012, 98, 917–921.
- Zhao, Y.; Zheng, Y.; Liu, X.; Luo, Q.; Wu, D.; Liu, X.; Zou, L. Inhibiting trophoblast PAR-1 overexpression suppresses sFlt-1-induced anti-angiogenesis and abnormal vascular remodeling: A possible therapeutic approach for preeclampsia. Mol. Hum. Reprod. 2018, 24, 158–169.
- Montagnana, M.; Lippi, G.; Albiero, A.; Scevarolli, S.; Salvagno, G.L.; Franchi, M.; Guidi, G.C. Evaluation of metalloproteinases 2 and 9 and their inhibitors in physiologic and pre-eclamptic pregnancy. J. Clin. Lab. Anal. 2009, 23, 88–92.
- Li, W.; Mata, K.M.; Mazzuca, M.Q.; Khalil, R.A. Altered matrix metalloproteinase-2 and -9 expression/activity links placental ischemia and anti-angiogenic sFlt-1 to uteroplacental and vascular remodeling and collagen deposition in hypertensive pregnancy. Biochem. Pharmacol. 2014, 89, 370–385.
- Raffetto, J.D.; Khalil, R.A. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem. Pharmacol. 2007, 75, 346–359.
- Chen, J.; Khalil, R.A. Matrix Metalloproteinases in Normal Pregnancy and Preeclampsia. In Progress in Molecular Biology and Translational Science; Elsevier Academic Press: Cambridge, MA, USA, 2017; Volume 148, pp. 87–165.
- Suman, P.; Gupta, S.K. Comparative analysis of the invasion-associated genes expression pattern in first trimester trophoblastic (HTR-8/SVneo) and JEG-3 choriocarcinoma cells. Placenta 2012, 33, 874–877.
- Su, M.-T.; Tsai, P.-Y.; Tsai, H.-L.; Chen, Y.-C.; Kuo, P.-L. miR-346 and miR-582-3p-regulated EG-VEGF expression and trophoblast invasion via matrix metalloproteinases 2 and 9. BioFactors 2016, 43, 210–219.
- Isaka, K.; Usuda, S.; Ito, H.; Sagawa, Y.; Nakamura, H.; Nishi, H.; Suzuki, Y.; Li, Y.; Takayama, M. Expression and activity of matrix metalloproteinase 2 and 9 in human trophoblasts. Placenta 2003, 24, 53–64.
- Dang, Y.; Li, W.; Tran, V.; Khalil, R.A. EMMPRIN-mediated induction of uterine and vascular matrix metalloproteinases during pregnancy and in response to estrogen and progesterone. Biochem. Pharmacol. 2013, 86, 734–747.
- Zhu, J.-Y.; Pang, Z.-J.; Yu, Y.-H. Regulation of Trophoblast Invasion: The Role of Matrix Metalloproteinases. Rev. Obstet. Gynecol. 2012, 5, e137–e143.
- Otun, H.A.; Lash, G.; Innes, B.A.; Bulmer, J.N.; Naruse, K.; Hannon, T.; Searle, R.F.; Robson, S.C. Effect of tumour necrosis factor-α in combination with interferon-γ on first trimester extravillous trophoblast invasion. J. Reprod. Immunol. 2011, 88, 1–11.
- Lash, G.; Otun, H.A.; Innes, B.A.; Kirkley, M.; De Oliveira, L.; Searle, R.F.; Robson, S.C.; Bulmer, J.N. Interferon-γ inhibits extravillous trophoblast cell invasion by a mechanism that involves both changes in apoptosis and protease levels. FASEB J. 2006, 20, 2512–2518.
- Choi, S.; Kim, J.A.; Li, H.-Y.; Lee, S.-J.; Seok, Y.S.; Kim, T.H.; Han, K.-H.; Park, M.H.; Cho, G.J.; Suh, S.H. Altered Redox State Modulates Endothelial KCa2.3 and KCa3.1 Levels in Normal Pregnancy and Preeclampsia. Antioxidants Redox Signal. 2019, 30, 505–519.
- Brähler, S.; Kaistha, A.; Schmidt, V.J.; Wölfle, S.E.; Busch, C.; Kaistha, B.P.; Kacik, M.; Hasenau, A.-L.; Grgic, I.; Si, H.; et al. Genetic Deficit of SK3 and IK1 Channels Disrupts the Endothelium-Derived Hyperpolarizing Factor Vasodilator Pathway and Causes Hypertension. Circulation 2009, 119, 2323–2332.
- Si, H.; Heyken, W.-T.; Wölfle, S.E.; Tysiac, M.; Schubert, R.; Grgic, I.; Vilianovich, L.; Giebing, G.; Maier, T.; Gross, V.; et al. Impaired Endothelium-Derived Hyperpolarizing Factor-Mediated Dilations and Increased Blood Pressure in Mice Deficient of the Intermediate-Conductance Ca 2+ -Activated K + Channel. Circ. Res. 2006, 99, 537–544.
- Barneo-Caragol, C.; Martínez-Morillo, E.; Rodríguez-González, S.; Lequerica-Fernández, P.; Vega-Naredo, I.; Alvarez, F.V. Increased serum strontium levels and altered oxidative stress status in early-onset preeclampsia. Free Radic. Biol. Med. 2019, 138, 1–9.
- Campbell, N.; Lamarca, B.D.; Cunningham, M.W. The Role of Agonistic Autoantibodies to the Angiotensin II Type 1 Receptor (AT1-AA) in Pathophysiology of Preeclampsia. Curr. Pharm. Biotechnol. 2018, 19, 781–785.
- Murphy, S.R.; Cockrell, K. Regulation of soluble fms-like tyrosine kinase-1 production in response to placental ischemia/hypoxia: Role of angiotensin II. Physiol. Rep. 2015, 3.
- El-Saka, M.H.; Madi, N.M.; Ibrahim, R.R.; Alghazaly, G.M.; Elshwaikh, S.; El-Bermawy, M. The ameliorative effect of angiotensin 1-7 on experimentally induced-preeclampsia in rats: Targeting the role of peroxisome proliferator-activated receptors gamma expression & asymmetric dimethylarginine. Arch. Biochem. Biophys. 2019, 671, 123–129.
- Wheeler, K.C.; Jena, M.; Pradhan, B.; Nayak, N.; Das, S.; Hsu, C.-D.; Wheeler, D.S.; Chen, K.; Nayak, N.R. VEGF may contribute to macrophage recruitment and M2 polarization in the decidua. PLoS ONE 2018, 13, e0191040.
- Jena, M.; Nayak, N.; Chen, K.; Nayak, N.R. Role of Macrophages in Pregnancy and Related Complications. Arch. Immunol. Ther. Exp. 2019, 67, 295–309.
- Yang, X.; Yang, Y.; Yuan, Y.; Liu, L.; Meng, T. The Roles of Uterine Natural Killer (NK) Cells and KIR/HLA-C Combination in the Development of Preeclampsia: A Systematic Review. BioMed Res. Int. 2020, 2020, 4808072-10.
- Blois, S.; Klapp, B.F.; Barrientos, G. Decidualization and angiogenesis in early pregnancy: Unravelling the functions of DC and NK cells. J. Reprod. Immunol. 2011, 88, 86–92.
- Le Bouteiller, P.; Tabiasco, J. Killers become builders during pregnancy. Nat. Med. 2006, 12, 991–992.
- Le Bouteiller, P.; Piccinni, M.-P. REVIEW ARTICLE: Human NK Cells in Pregnant Uterus: Why There? Am. J. Reprod. Immunol. 2008, 59, 401–406.
- Tayade, C.; Hilchie, D.; He, H.; Fang, Y.; Moons, L.; Carmeliet, P.; Foster, R.A.; Croy, B.A. Genetic deletion of placenta growth factor in mice alters uterine NK cells. J. Immunol. 2007, 178, 4267–4275.
- Hanna, J.H.; Goldman-Wohl, D.; Hamani, Y.; Avraham, I.; Greenfield, C.; Natanson-Yaron, S.; Prus, D.; Cohen-Daniel, L.; Arnon, T.I.; Manaster, I.; et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat. Med. 2006, 12, 1065–1074.
- Szarka, A.; Rigó, J.; Lázár, L.; Beko, G.; Molvarec, A. Circulating cytokines, chemokines and adhesion molecules in normal pregnancy and preeclampsia determined by multiplex suspension array. BMC Immunol. 2010, 11, 59.
- Gotsch, F.; Romero, R.; Friel, L.; Kusanovic, J.P.; Espinoza, J.; Erez, O.; Than, N.G.; Mittal, P.; Edwin, S.; Yoon, B.H.; et al. CXCL10/IP-10: A missing link between inflammation and anti-angiogenesis in preeclampsia? J. Matern. Neonatal Med. 2007, 20, 777–792.
- Du, M.-R.; Wang, S.-C.; Li, D.-J. The integrative roles of chemokines at the maternal–fetal interface in early pregnancy. Cell. Mol. Immunol. 2014, 11, 438–448.
- Fraser, R.; Whitley, G.S.; Johnstone, A.P.; Host, A.J.; Sebire, N.J.; Thilaganathan, B.; Cartwright, J.E. Impaired decidual natural killer cell regulation of vascular remodelling in early human pregnancies with high uterine artery resistance. J. Pathol. 2012, 228, 322–332.
- Wallace, A.E.; Host, A.J.; Whitley, G.S.; Cartwright, J.E. Decidual natural killer cell interactions with trophoblasts are impaired in pregnancies at increased risk of preeclampsia. Am. J. Pathol. 2013, 183, 1853–1861.
- Harmon, A.C.; Ibrahim, T.; Cornelius, D.C.; Amaral, L.M.; Cunningham, M.W.; Wallace, K.; Lamarca, B.D. Placental CD4+ T cells isolated from preeclamptic women cause preeclampsia-like symptoms in pregnant nude-athymic rats. Pregnancy Hypertens. 2018, 15, 7–11.
- Herse, F.; Lamarca, B. Angiotensin II type 1 receptor autoantibody (AT1-AA)-mediated pregnancy hypertension. Am. J. Reprod. Immunol. 2012, 69, 413–418.
- Weel, I.C.; Baergen, R.N.; Veiga, M.R.; Borges, V.; Ribeiro, V.R.; Witkin, S.S.; Castro, C.F.B.; Peraçoli, J.C.; De Oliveira, L.; Peracoli, M.T. Association between Placental Lesions, Cytokines and Angiogenic Factors in Pregnant Women with Preeclampsia. PLoS ONE 2016, 11, e0157584.
- Parchim, N.F.; Wang, W.; Iriyama, T.; Ashimi, O.A.; Siddiqui, A.H.; Blackwell, S.; Sibai, B.; Kellems, R.E.; Xia, Y. Neurokinin 3 receptor and phosphocholine transferase: Missing factors for pathogenesis of C-reactive protein in preeclampsia. Hypertension 2014, 65, 430–439.
- Raio, L.; Bersinger, N.A.; Malek, A.; Schneider, H.; Messerli, F.H.; Hürter, H.; Rimoldi, S.F.; Baumann, M.U. Ultra-high sensitive C-reactive protein during normal pregnancy and in preeclampsia. J. Hypertens. 2019, 37, 1012–1017.
- Penning, M.; Chua, J.S.; Van Kooten, C.; Zandbergen, M.; Buurma, A.; Schutte, J.; Bruijn, J.A.; Khankin, E.; Bloemenkamp, K.; Karumanchi, S.A.; et al. Classical Complement Pathway Activation in the Kidneys of Women With Preeclampsia. Hypertension 2015, 66, 117–125.
- Burwick, R.M.; Fichorova, R.N.; Dawood, H.Y.; Yamamoto, H.S.; Feinberg, B.B. Urinary Excretion of C5b-9 in Severe Preeclampsia. Hypertension 2013, 62, 1040–1045.
- Burwick, R.M.; Velasquez, J.; Valencia, C.M.; Gutiérrez-Marín, J.; Edna-Estrada, F.; Silva, J.L.; Trujillo-Otálvaro, J.; Vargas-Rodríguez, J.; Bernal, Y.; Quintero, A.; et al. Terminal Complement Activation in Preeclampsia. Obstet. Gynecol. 2018, 132, 1477–1485.
- Ma, Y.; Kong, L.; Ge, Q.; Lu, Y.; Hong, M.; Zhang, Y.; Ruan, C.-C.; Gao, P. Complement 5a-mediated trophoblasts dysfunction is involved in the development of pre-eclampsia. J. Cell. Mol. Med. 2017, 22, 1034–1046.
- Brož, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420.
- Petrilli, V.; Papin, S.; Tschopp, J. The inflammasome. Curr. Biol. 2005, 15, R581.
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241.
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Núñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361.
- Stødle, G.S.; Silva, G.B.; Tangerås, L.H.; Gierman, L.M.; Nervik, I.; Dahlberg, U.E.; Sun, C.; Aune, M.H.; Thomsen, L.C.V.; Bjørge, L.; et al. Placental inflammation in pre-eclampsia by Nod-like receptor protein (NLRP)3 inflammasome activation in trophoblasts. Clin. Exp. Immunol. 2018, 193, 84–94.
- Robillard, P.-Y.; Dekker, G.; Hulsey, T.C. Revisiting the epidemiological standard of preeclampsia: Primigravidity or primipaternity? Eur. J. Obstet. Gynecol. Reprod. Biol. 1999, 84, 37–41.
- Deen, M.E.; Ruurda, L.G.C.; Wang, J.; Dekker, G. Risk factors for preeclampsia in multiparous women: Primipaternity versus the birth interval hypothesis. J. Matern. Neonatal Med. 2006, 19, 79–84.
- Heim, K.; Mulla, M.J.; Potter, J.A.; Han, C.S.; Guller, S.; Abrahams, V.M. Excess glucose induce trophoblast inflammation and limit cell migration through HMGB1 activation of Toll-Like receptor 4. Am. J. Reprod. Immunol. 2018, 80, e13044.
- Kweider, N.; Wruck, C.J.; Rath, W. New Insights into the Pathogenesis of Preeclampsia—The Role of Nrf2 Activators and their Potential Therapeutic Impact. Geburtshilfe und Frauenheilkd 2013, 73, 1236–1240.
- Kweider, N.; Huppertz, B.; Kadyrov, M.; Rath, W.; Pufe, T.; Wruck, C. A possible protective role of Nrf2 in preeclampsia. Ann. Anat. Anat. Anz. 2014, 196, 268–277.
- Zeng, X.; Huang, Z.; Mao, X.; Wang, J.; Wu, G.; Qiao, S. N-Carbamylglutamate Enhances Pregnancy Outcome in Rats through Activation of the PI3K/PKB/mTOR Signaling Pathway. PLoS ONE 2012, 7, e41192.
- Wu, D.; Hong, H.; Huang, X.; Huang, L.; He, Z.; Fang, Q.; Luo, Y. CXCR2 is decreased in preeclamptic placentas and promotes human trophoblast invasion through the Akt signaling pathway. Placenta 2016, 43, 17–25.
- Lai, W.; Ding, Y. GNG7 silencing promotes the proliferation and differentiation of placental cytotrophoblasts in preeclampsia rats through activation of the mTOR signaling pathway. Int. J. Mol. Med. 2019, 43, 1939–1950.
- Lin, L.; Li, G.; Zhang, W.; Wang, Y.; Yang, H. Low-dose aspirin reduces hypoxia-induced sFlt1 release via the JNK/AP-1 pathway in human trophoblast and endothelial cells. J. Cell. Physiol. 2019, 234, 18928–18941.
- Munaut, C.; Tebache, L.; Blacher, S.; Noel, A.; Nisolle, M.; Chantraine, F. Dysregulated circulating miRNAs in preeclampsia. Biomed. Rep. 2016, 5, 686–692.
- Jiang, W.-L.; Zhang, Y.; Xia, Q.; Zhu, J.; Yu, X.; Fan, T.; Wang, F. MicroRNA-19a regulates lipopolysaccharide-induced endothelial cell apoptosis through modulation of apoptosis signal-regulating kinase 1 expression. BMC Mol. Biol. 2015, 16, 11.
- Zhang, J.; Xiao, Z.; Lai, D.; Sun, J.; He, C.; Chu, Z.; Ye, H.; Chen, S.; Wang, J. miR-21, miR-17 and miR-19a induced by phosphatase of regenerating liver-3 promote the proliferation and metastasis of colon cancer. Br. J. Cancer 2012, 107, 352–359.
- Müller-Deile, J.; Schröder, P.; Beverly-Staggs, L.; Hiss, R.; Fiedler, J.; Nystrom, J.; Thum, T.; Haller, H.; Schiffer, M. Overexpression of preeclampsia induced microRNA-26a-5p leads to proteinuria in zebrafish. Sci. Rep. 2018, 8, 3621.
- Sandrim, V.; Eleuterio, N.; Pilan, E.; Tanus-Santos, J.E.; Fernandes, K.; Cavalli, R.D.C. Plasma levels of increased miR-195-5p correlates with the sFLT-1 levels in preeclampsia. Hypertens. Pregnancy 2016, 35, 1–9.
- Moore, T.; Dveksler, G. Pregnancy-specific glycoproteins: Complex gene families regulating maternal-fetal interactions. Int. J. Dev. Biol. 2014, 58, 273–280.
- Wang, N.; Li, R.; Xue, M. Potential regulatory network in the PSG10P/miR-19a-3p/IL1RAP pathway is possibly involved in preeclampsia pathogenesis. J. Cell. Mol. Med. 2018, 23, 852–864.
- Tsialikas, J.; Romer-Seibert, J. LIN28: Roles and regulation in development and beyond. Development 2015, 142, 2397–2404.
- Canfield, J.; Arlıer, S.; Mong, E.F.; Lockhart, J.; Van Wye, J.; Guzeloglu-Kayisli, O.; Schatz, F.; Magness, R.R.; Lockwood, C.J.; Tsibris, J.C.M.; et al. Decreased LIN28B in preeclampsia impairs human trophoblast differentiation and migration. FASEB J. 2018, 33, 2759–2769.
- Miura, K.; Higashijima, A.; Murakami, Y.; Tsukamoto, O.; Hasegawa, Y.; Abe, S.; Fuchi, N.; Miura, S.; Kaneuchi, M.; Masuzaki, H. Circulating chromosome 19 miRNA cluster microRNAs in pregnant women with severe pre-eclampsia. J. Obstet. Gynaecol. Res. 2015, 41, 1526–1532.
- Wapinski, O.; Chang, H.Y. Long noncoding RNAs and human disease. Trends Cell Biol. 2011, 21, 354–361.
- He, X.; He, Y.; Xi, B.; Zheng, J.; Zeng, X.; Cai, Q.; Ouyang, Y.; Wang, C.; Zhou, X.; Huang, H.; et al. LncRNAs Expression in Preeclampsia Placenta Reveals the Potential Role of LncRNAs Contributing to Preeclampsia Pathogenesis. PLoS ONE 2013, 8, e81437.
- Wagner, O.F.; Christ, G.; Wojta, J.; Vierhapper, H.; Parzer, S.; Nowotny, P.J.; Schneider, B.; Waldhäusl, W.; Binder, B.R. Polar secretion of endothelin-1 by cultured endothelial cells. J. Biol. Chem. 1992, 267, 16066–16068.
- Galiè, N.; Manes, A.; Branzi, A. The endothelin system in pulmonary arterial hypertension. Cardiovasc. Res. 2004, 61, 227–237.
- Granger, J.; Spradley, F.T.; Bakrania, B.A. The Endothelin System: A Critical Player in the Pathophysiology of Preeclampsia. Curr. Hypertens. Rep. 2018, 20, 32.
- Bakrania, B.A.; Duncan, J.; Warrington, J.P.; Granger, J.P. The Endothelin Type A Receptor as a Potential Therapeutic Target in Preeclampsia. Int. J. Mol. Sci. 2017, 18, 522.
- Saleh, L.; Verdonk, K.; Visser, W.; Meiracker, A.H.V.D.; Danser, A.J. The emerging role of endothelin-1 in the pathogenesis of pre-eclampsia. Ther. Adv. Cardiovasc. Dis. 2016, 10, 282–293.
- Tomimatsu, T.; Mimura, K.; Matsuzaki, S.; Endo, M.; Kumasawa, K.; Kimura, T. Preeclampsia: Maternal Systemic Vascular Disorder Caused by Generalized Endothelial Dysfunction Due to Placental Antiangiogenic Factors. Int. J. Mol. Sci. 2019, 20, 4246.
- Shahul, S.; Tung, A.; Minhaj, M.; Nizamuddin, J.; Wenger, J.; Mahmood, E.; Mueller, A.; Shaefi, S.; Scavone, B.; Kociol, R.; et al. Racial Disparities in Comorbidities, Complications, and Maternal and Fetal Outcomes in Women With Preeclampsia/eclampsia. Hypertens. Pregnancy 2015, 34, 506–515.
- Reidy, K.J.; Hjorten, R.C.; Simpson, C.L.; Rosenberg, A.Z.; Rosenblum, S.D.; Kovesdy, C.P.; Tylavsky, F.A.; Myrie, J.; Ruiz, B.L.; Haque, S.; et al. Fetal—Not Maternal—APOL1 Genotype Associated with Risk for Preeclampsia in Those with African Ancestry. Am. J. Hum. Genet. 2018, 103, 367–376.
- Bdolah, Y.; Palomaki, G.E.; Yaron, Y.; Bdolah-Abram, T.; Goldman, M.; Levine, R.J.; Sachs, B.P.; Haddow, J.E.; Karumanchi, S.A. Circulating angiogenic proteins in trisomy 13. Am. J. Obstet. Gynecol. 2006, 194, 239–245.
- Skjærven, R.; Vatten, L.J.; Wilcox, A.J.; Rønning, T.; Irgens, L.M.; Lie, R.T. Recurrence of pre-eclampsia across generations: Exploring fetal and maternal genetic components in a population based cohort. BMJ 2005, 331, 877.
- Esplin, M.S.; Fausett, M.; Fraser, A.; Kerber, R.; Mineau, G.; Carrillo, J.; Varner, M.W. Paternal and Maternal Components of the Predisposition to Preeclampsia. N. Engl. J. Med. 2001, 344, 867–872.
- McGinnis, R.; The FINNPEC Consortium; Steinthorsdottir, V.; Williams, N.O.; Thorleifsson, G.; Shooter, S.; Hjartardottir, S.; Bumpstead, S.; Stefansdottir, L.; Hildyard, L.; et al. Variants in the fetal genome near FLT1 are associated with risk of preeclampsia. Nat. Genet. 2017, 49, 1255–1260.
- Gray, K.J.; Saxena, R.; Karumanchi, S.A. Genetic predisposition to preeclampsia is conferred by fetal DNA variants near FLT1, a gene involved in the regulation of angiogenesis. Am. J. Obstet. Gynecol. 2018, 218, 211–218.
- Cole, L.A. Biological functions of hCG and hCG-related molecules. Reprod. Biol. Endocrinol. 2010, 8, 102.
- Barjaktarovic, M.; Korevaar, T.I.M.; Jaddoe, V.W.V.; Rijke, Y.B.; Peeters, R.P.; Steegers, E.A.; De Rijke, Y.B. Human chorionic gonadotropin and risk of pre-eclampsia: Prospective population-based cohort study. Ultrasound Obstet. Gynecol. 2019, 54, 477–483.
- Paiva, S.P.; Veloso, C.A.; Campos, F.F.; Carneiro, M.; Tilan, J.U.; Wang, H.; Umans, J.G.; Zukowska, Z.; Kitlinska, J. Elevated levels of neuropeptide Y in preeclampsia: A pilot study implicating a role for stress in pathogenesis of the disease. Neuropeptides 2015, 55, 127–135.
- Schröder, M.; Kaufman, R.J. The Mammalian Unfolded Protein Response. Annu. Rev. Biochem. 2005, 74, 739–789.
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529.
- Gerasimova, E.M.; Fedotov, S.A.; Kachkin, D.V.; Vashukova, E.; Glotov, A.; Chernoff, Y.O.; Rubel, A. Protein Misfolding during Pregnancy: New Approaches to Preeclampsia Diagnostics. Int. J. Mol. Sci. 2019, 20, 6183.
- Nakakita, B.; Mogami, H.; Kondoh, E.; Tsukamoto, T.; Yanagita, M.; Konishi, I. Case of soluble fms-like tyrosine kinase 1 apheresis in severe pre-eclampsia developed at 15 weeks’ gestation. J. Obstet. Gynaecol. Res. 2015, 41, 1661–1663.
- Yang, S.; Song, L.; Shi, X.; Zhao, N.; Ma, Y. Ameliorative effects of pre-eclampsia by quercetin supplement to aspirin in a rat model induced by L-NAME. Biomed. Pharmacother. 2019, 116, 108969.
- Kumasawa, K.; Ikawa, M.; Kidoya, H.; Hasuwa, H.; Saito-Fujita, T.; Morioka, Y.; Takakura, N.; Kimura, T.; Okabe, M. Pravastatin induces placental growth factor (PGF) and ameliorates preeclampsia in a mouse model. Proc. Natl. Acad. Sci. USA 2010, 108, 1451–1455.
- Costantine, M.M.; Tamayo, E.; Lu, F.; Bytautiene, E.; Longo, M.; Hankins, G.D.V.; Saade, G.R. Using Pravastatin to Improve the Vascular Reactivity in a Mouse Model of Soluble Fms-Like Tyrosine Kinase-1–Induced Preeclampsia. Obstet. Gynecol. 2010, 116, 114–120.
- Fox, K.; Longo, M.; Tamayo, E.; Kechichian, T.; Bytautiene, E.; Hankins, G.D.; Saade, G.; Costantine, M.M. Effects of pravastatin on mediators of vascular function in a mouse model of soluble Fms-like tyrosine kinase-1–induced preeclampsia. Am. J. Obstet. Gynecol. 2011, 205, 366.e1–366.e5.
- Saad, A.F.; Kechichian, T.; Yin, H.; Sbrana, E.; Longo, M.; Wen, M.; Tamayo, E.; Hankins, G.D.V.; Saade, G.R.; Costantine, M.M. Effects of Pravastatin on Angiogenic and Placental Hypoxic Imbalance in a Mouse Model of Preeclampsia. Reprod. Sci. 2013, 21, 138–145.
- Singh, J.; Ahmed, A.; Girardi, G. Role of Complement Component C1q in the Onset of Preeclampsia in Mice. Hypertension 2011, 58, 716–724.
- Kim, J.-Y.; Cho, H.-J.; Sir, J.-J.; Kim, B.-K.; Hur, J.; Youn, S.-W.; Yang, H.-M.; Jun, S.-I.; Park, K.W.; Hwang, S.-J.; et al. Sulfasalazine induces haem oxygenase-1 via ROS-dependent Nrf2 signalling, leading to control of neointimal hyperplasia. Cardiovasc. Res. 2009, 82, 550–560.
- Brownfoot, F.C.; Hannan, N.J.; Cannon, P.; Nguyen, V.; Hastie, R.; Parry, L.J.; Senadheera, S.; Tuohey, L.; Tong, S.; Kaitu’U-Lino, T.J. Sulfasalazine reduces placental secretion of antiangiogenic factors, up-regulates the secretion of placental growth factor and rescues endothelial dysfunction. EBioMedicine 2019, 41, 636–648.
- Santiago-Font, J.A.; Amaral, L.M.; Faulkner, J.; Ibrahim, T.; Vaka, V.R.; Cunningham, M.W.; Lamarca, B.D. Serelaxin improves the pathophysiology of placental ischemia in the reduced uterine perfusion pressure rat model of preeclampsia. Am. J. Physiol. Integr. Comp. Physiol. 2016, 311, R1158–R1163.
- Conrad, K.P.; Benyo, D.F. Placental Cytokines and the Pathogenesis of Preeclampsia. Am. J. Reprod. Immunol. 1997, 37, 240–249.
- Lamarca, B. The role of immune activation in contributing to vascular dysfunction and the pathophysiology of hypertension during preeclampsia. Minerva Ginecol. 2010, 62, 105–120.
- Matsubara, K.; Matsubara, Y.; Hyodo, S.; Katayama, T.; Ito, M. Role of nitric oxide and reactive oxygen species in the pathogenesis of preeclampsia. J. Obstet. Gynaecol. Res. 2010, 36, 239–247.
- Sandrim, V.; Montenegro, M.F.; Palei, A.C.; Metzger, I.F.; Sertório, J.T.; Cavalli, R.D.C.; Tanus-Santos, J.E. Increased circulating cell-free hemoglobin levels reduce nitric oxide bioavailability in preeclampsia. Free Radic. Biol. Med. 2010, 49, 493–500.
- Conrad, K.P. Emerging Role of Relaxin in the Maternal Adaptations to Normal Pregnancy: Implications for Preeclampsia. Semin. Nephrol. 2011, 31, 15–32.
- Sasser, J.M. The emerging role of relaxin as a novel therapeutic pathway in the treatment of chronic kidney disease. Am. J. Physiol. Integr. Comp. Physiol. 2013, 305, R559–R565.
- Shi, J.; Wei, L. Rho kinases in cardiovascular physiology and pathophysiology: The effect of fasudil. J. Cardiovasc. Pharmacol. 2013, 62, 341–354.
- Gu, Y.; Feng, Y.; Yu, J.; Yuan, H.; Yin, Y.; Ding, J.; Zhao, J.; Xu, Y.; Xu, J.; Che, H. Fasudil attenuates soluble fms-like tyrosine kinase-1 (sFlt-1)-induced hypertension in pregnant mice through RhoA/ROCK pathway. Oncotarget 2017, 8, 104104–104112.
- Wojciak-Stothard, B.; Tsang, L.Y.F.; Haworth, S.G. Rac and Rho play opposing roles in the regulation of hypoxia/reoxygenation-induced permeability changes in pulmonary artery endothelial cells. Am. J. Physiol. Cell. Mol. Physiol. 2005, 288, L749–L760.
- Zhou, L.; Qiao, F. Expression of RhoA in placenta of preeclampsia. Acta Acad. Med. Wuhan 2006, 26, 744–746.
- Liu, X.; Deng, Q.; Luo, X.; Chen, Y.; Shan, N.; Qi, H. Oxidative stress-induced Gadd45α inhibits trophoblast invasion and increases sFlt1/sEng secretions via p38 MAPK involving in the pathology of pre-eclampsia. J. Matern. Neonatal Med. 2016, 29, 3776–3785.
- Feng, Y.; Hu, L.; Xu, Q.; Yuan, H.; Ba, L.; He, Y.; Che, H. Cytoprotective Role of Alpha-1 Antitrypsin in Vascular Endothelial Cell Under Hypoxia/Reoxygenation Condition. J. Cardiovasc. Pharmacol. 2015, 66, 96–107.
- Eddy, A.C.; Bidwell, G.L.; George, E.M. Pro-angiogenic therapeutics for preeclampsia. Biol. Sex Differ. 2018, 9, 36.
- Romero, R.; Erez, O.; Hüttemann, M.; Maymon, E.; Panaitescu, B.; Conde-Agudelo, A.; Pacora, P.; Yoon, B.H.; Grossman, L.I. Metformin, the aspirin of the 21st century: Its role in gestational diabetes mellitus, prevention of preeclampsia and cancer, and the promotion of longevity. Am. J. Obstet. Gynecol. 2017, 217, 282–302.
- Putra, R.A.; Effendi, J.S.; Permadi, W.; Bandiara, R.; Fauziah, P.N. Role of statin as inducer of Hmox-1 system in treatment of preeclampsia. Cell. Mol. Biol. 2018, 64, 1–4.
- Song, J.; Li, Y.; An, R. Vitamin D restores angiogenic balance and decreases tumor necrosis factor-α in a rat model of pre-eclampsia. J. Obstet. Gynaecol. Res. 2016, 43, 42–49.
- Ushida, T.; Kotani, T.; Tsuda, H.; Imai, K.; Nakano, T.; Hirako, S.; Ito, Y.; Li, H.; Mano, Y.; Wang, J.; et al. Molecular hydrogen ameliorates several characteristics of preeclampsia in the Reduced Uterine Perfusion Pressure (RUPP) rat model. Free Radic. Biol. Med. 2016, 101, 524–533.
- Carver, A.R.; Andrikopoulou, M.; Lei, J.; Tamayo, E.; Gamble, P.; Hou, Z.; Zhang, J.; Mori, S.; Saade, G.R.; Costantine, M.M.; et al. Maternal Pravastatin Prevents Altered Fetal Brain Development in a Preeclamptic CD-1 Mouse Model. PLoS ONE 2014, 9, e100873.
- McDonnold, M.; Tamayo, E.; Kechichian, T.; Gamble, P.; Longo, M.; Hankins, G.D.; Saade, G.; Costantine, M.M. The effect of prenatal pravastatin treatment on altered fetal programming of postnatal growth and metabolic function in a preeclampsia-like murine model. Am. J. Obstet. Gynecol. 2014, 210, 542.e1–542.e7.
- Saad, A.F.; Diken, Z.M.; Kechichian, T.B.; Clark, S.M.; Olson, G.L.; Saade, G.R.; Costantine, M.M. Pravastatin Effects on Placental Prosurvival Molecular Pathways in a Mouse Model of Preeclampsia. Reprod. Sci. 2016, 23, 1593–1599.
- Zhao, Y.; Zheng, Y.; Luo, Q.; Yan, T.; Liu, X.; Han, L.; Zou, L. Edaravone inhibits hypoxia-induced trophoblast-soluble Fms-like tyrosine kinase 1 expression: A possible therapeutic approach to preeclampsia. Placenta 2014, 35, 476–482.



