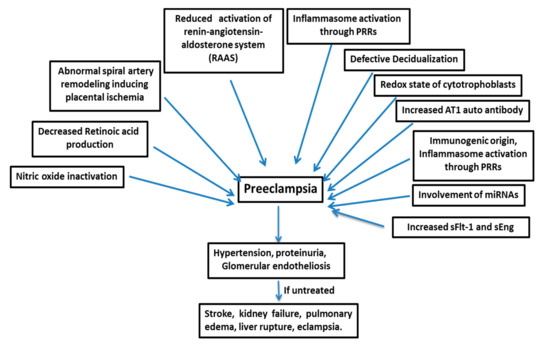
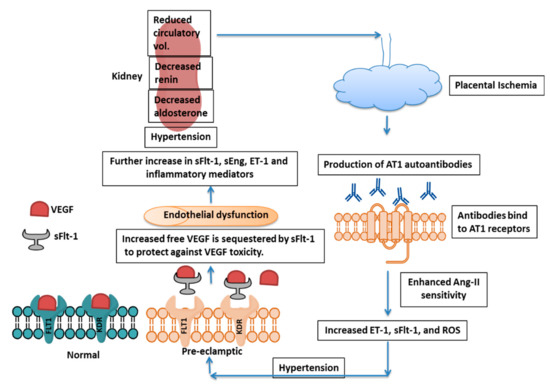
3. Therapeutic Approaches
Various therapeutic reagents have been predicted to be effective in managing the preeclamptic condition, although delivery of the placenta is the only solution to relieve maternal hypertension and proteinuria.
In a human case of very early-onset PE (15 weeks of gestation), sFlt-1 apheresis was performed 13 times in of the interval from 19 to 23 weeks of gestation; however, at 23 weeks and 3 days-cesarean section had to be performed due to maternal respiratory failure and fetal distress. This study indicates sFlt-1 functions to protect the placenta and fetus in PE and that removal may have negative consequences
[170][180].
A study in a rat model of PE induced by NG-nitro-Larginine-methyl ester (L-NAME) to evaluate the therapeutic effect of quercetin (a bioflavonoid having antioxidant and reno-protective properties) in combination with aspirin showed amelioration of symptoms through a reduction in sFlt-1 and VEGF levels in the uterus
[171][181]. Moreover, the prenatal treatment of preeclamptic animal models with pravastatin (a drug of choice to lower bad cholesterol) improved blood pressure, vascular activity, and pup growth and led to an increase in VEGF and PlGF levels and a decrease in sFlt-1 levels
[172][173][174][175][176][179,182,183,184,185].
The drug sulfasalazine (an anti-inflammatory and antioxidant), used to treat autoimmune diseases and found to be safe during pregnancy
[177][186] was shown to reduce sFlt-1 and sEng levels and increase PlGF secretion from human placenta
[178][187]. Additionally, sulfasalazine, which mitigates endothelial dysfunction, a major pathologic condition in PE could be used as a therapeutic agent for PE but needs further investigation. A study to determine the therapeutic efficacy of relaxin (serelaxin, i.e., recombinant human relaxin-2) for the treatment of PE revealed this hormone reduces blood pressure and the levels of circulating TNF-α, sFlt-1, and preproendothelin while simultaneously increasing NO bioavailability in RUPP rats
[179][188]. In PE, there is a decreased level of vasodilators, like NO, and increased levels of vasoconstrictors, like ET-1
[73][180][181][182][183][73,189,190,191,192]. Relaxin is a protein hormone of 6 kDa MW produced by ovaries (corpora lutea), cells of non-pregnant endometrium, decidual cells of pregnant endometrium, and blood vessels, cytotrophoblasts, and syncytiotrophoblasts
[184][193]. In addition to its role in the relaxation of skeletal soft tissue and the cardiovascular and renal systems, relaxin plays an important role in maintaining blood pressure in normal pregnancy and is found to reduce blood pressure in a rodent model of hypertension
[185][194].
Fasudil is a first-generation Rho/Rho-associated protein kinase (ROCK) inhibitor frequently used for the treatment of hypertension and other cardiovascular diseases
[186][195]. A study by Gu et al. (2017) revealed that fasudil can attenuate hypertension induced by sFlt-1 in preeclamptic mice through inhibition of the RhoA/ROCK pathway
[187][196]. Rho GTPases play crucial roles coupling the cellular redox state to endothelial cell function
[188][197]. RhoA (Ras homolog gene family, member A) proteins are expressed at higher levels in PE, suggesting a role in PE pathogenesis
[189][198]. Antioxidants like vit-C and vit-E inhibit the p38 signaling pathway and thus block sFlt-1 secretion in hypoxia-reoxygenation-induced endothelial cell monolayers
[190][199].
Studies have also revealed that exogenous alpha-1 anti-trypsin can alleviate hypoxia/reoxygenation injury by reducing oxidative stress through inactivation of Rac1/p38 signaling
[191][200]. Eddy et al. (2018) reviewed the use of VEGF and PlGF as therapeutics to curb PE and suggested the modified stabilized members of the VEGF family could be used as therapeutic agents for treatment
[192][201], but recent studies reveal VEGF and PlGF could be triggers for increased sFlt-1 production. A study of metformin showed it was able to prevent PE, reducing the production of sFlt-1 and sEng and ameliorating endothelial dysfunction through effects on mitochondria
[193][202]. The protein statin exerts a protective effect on endothelial cells through induction of Hmox-1 expression and inhibition of sFlt-1 release, along with its antioxidant properties
[194][203].
Vitamin D has a therapeutic effect on PE, as observed in the l-nitro-arginine methyl ester-induced PE rat models. Vitamin D supplementation was found to increase VEGF levels and decrease sFlt-1 and TNF-α levels in PE rats. Vitamin D also reduced oxidative stress by lowering the levels of malondialdehyde (plasma oxidative stress marker)
[195][204]. Moreover, molecular hydrogen (H
2) has therapeutic effects in several oxidative stress-related disorders. In the RUPP rat model of PE, H2 reduced mean arterial pressure and sFlt-1 expression. H2 also reduced sFlt-1 expression in villous explants taken from preeclamptic women. These studies show the preventive and therapeutic effects of H
2 on PE
[196][205].
A study with a CD-1 mouse model of PE showed maternal treatment with pravastatin prevents alterations in fetal brain development, growth, and metabolic functions
[197][198][206,207]. It has been shown that PE alters brain development in sex-specific patterns, and prenatal pravastatin therapy prevents changes in neuroanatomic programming that occur in the preeclamptic CD-1 mouse model
[197][206]. Pravastatin may exert its effects through pleiotropic mechanisms involving the pro-survival/antiapoptotic MAPK pathway in the placenta
[199][208]. One more drug, edaravone (free radical scavenger), has inhibitory effects on the expression of sFlt-1 in the hypoxia-induced HTR8/SVneo trophoblast cell line. This compound showed a protective effect on the vascular development of human umbilical vein endothelial cells (HUVECs) in hypoxia, proving it to be a potential therapeutic agent for PE treatment
[200][209]. Experimental animals administered exogenous VEGF121 also show an alleviation of PE symptoms
[46][47].