Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | P. Syamasundar Rao | + 4739 word(s) | 4739 | 2021-05-31 08:10:35 | | | |
| 2 | Bruce Ren | -21 word(s) | 4718 | 2021-06-08 05:12:09 | | | | |
| 3 | Bruce Ren | Meta information modification | 4718 | 2021-06-10 07:53:24 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Rao, P.S. Single Ventricle. Encyclopedia. Available online: https://encyclopedia.pub/entry/10601 (accessed on 23 July 2026).
Rao PS. Single Ventricle. Encyclopedia. Available at: https://encyclopedia.pub/entry/10601. Accessed July 23, 2026.
Rao, P. Syamasundar. "Single Ventricle" Encyclopedia, https://encyclopedia.pub/entry/10601 (accessed July 23, 2026).
Rao, P.S. (2021, June 07). Single Ventricle. In Encyclopedia. https://encyclopedia.pub/entry/10601
Rao, P. Syamasundar. "Single Ventricle." Encyclopedia. Web. 07 June, 2021.
Copy Citation
In this paper, the author enumerates cardiac defects with a functionally single ventricle, summarizes single ventricle physiology, presents a summary of management strategies to address the single ventricle defects, goes over the steps of staged total cavo-pulmonary connection, cites the prevalence of inter-stage mortality, names the causes of inter-stage mortality, discusses strategies to address the inter-stage mortality, reviews post-Fontan issues, and introduces alternative approaches to Fontan circulation.
single ventricle
double-inlet left ventricle
hypoplastic left heart syndrome
tricuspid atresia
unbalanced atrioventricular septal defect
mitral atresia with normal aortic root
heterotaxy syndromes
Down syndrome
Fontan circulation
biventricular repair
1. Introduction
The term “single ventricle” is generally utilized to describe any congenital heart defect (CHD) with one functioning ventricle, and these are: double-inlet left ventricle (DILV), single ventricle, common ventricle, and univentricular atrio-ventricular (AV) connection [1]. Other lesions, namely hypoplastic left heart syndrome (HLHS), tricuspid atresia, unbalanced AV septal defect, mitral atresia with normal aortic root, and heterotaxy syndromes with one functioning ventricle may now be added to this group.
2. Cardiac Defects with Functionally Single Ventricle
As mentioned above, there are number of cardiac defects that have a functionally single ventricle and are candidates for single ventricle/Fontan repair. These will be described briefly. The order of presentation is arbitrary.
2.1. Hypoplastic Left Heart Syndrome
The phrase “HLHS” was first suggested by Noonan and Nadas [2] to characterize a very small left ventricle with poorly developed aortic and mitral valves. The left ventricle (LV) is usually a slit-like cavity (Figure 1) with thick muscle, especially when mitral atresia co-exists. The aortic valve is atretic or markedly narrowed with annular hypoplasia. Similarly, the mitral valve is markedly stenotic, hypoplastic or atretic (Figure 1). The LV is very small when the mitral valve is open. Endocardial fibroelastosis is often present. Hypoplasia of the ascending aorta is present, and its diameter is usually 2–3 mm (Figure 2). However, it is adequate to supply ample coronary blood flow retrogradely. The left atrium is very small (Figure 1). The interatrial septum is usually thickened with a small patent foramen ovale (PFO) and rarely the atrial septum is intact. A patent ductus arteriosus (PDA) is classically present and is necessary for the baby to survive [3][4][5].
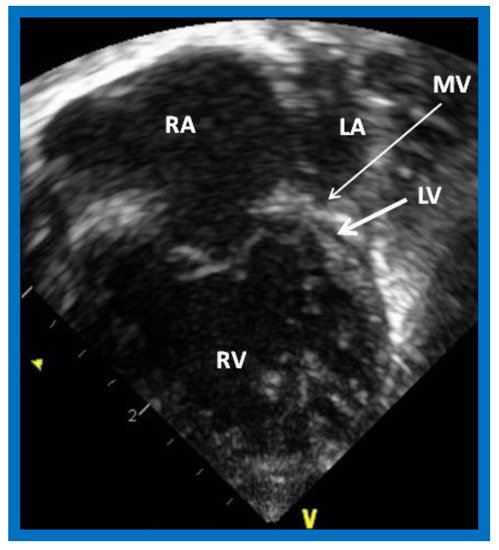
Figure 1. Echocardiogram in an apical 4-chamber view of a baby with hypoplastic left heart syndrome, showing a strikingly small, slit-like (thick arrow) left ventricle (LV), obviously enlarged and hypertrophied right ventricle (RV), and a dilated right atrium (RA). The atretic mitral valve (MV) (thin arrow) and hypoplastic left atrium (LA) are also seen.
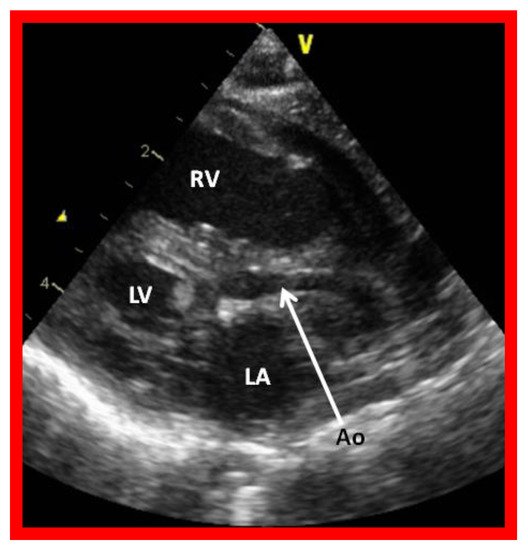
Figure 2. Echocardiogram in a parasternal long short axis view of a baby with hypoplastic left heart syndrome illustrates a small left ventricle (LV), a severely hypoplastic aorta (Ao) (arrow), and an enlarged right ventricle (RV). LA, left atrium.
2.2. Tricuspid Atresia
Tricuspid atresia (TA) is a cyanotic CHD and is characterized as a congenital absence or agenesis of the morphologic tricuspid valve [6][7]. The right atrium is dilated and the tricuspid valve is atretic (Figure 3). In the most common muscular variety, atretic tricuspid valve is seen as a localized fibrous thickening or a dimple in the floor of the right atrium at the anticipated site of the tricuspid valve [8][9][10][11][12]. An atrial septal defect is necessary for survival and is typically a stretched PFO. The mitral valve is usually bicuspid and morphologically a mitral valve. The LV is evidently a morphological LV, but it is enlarged and hypertrophied [8][9][10][11][12]. Usually, a ventricular septal defect (VSD) is present. The VSD is most commonly in the muscular ventricular septum [13][14]. The right ventricle (RV) is hypoplastic and is not sufficiently large in size to support pulmonary circulation. The origin of great arteries is variable and on the basis of which a classification of this disease entity was developed [6]: Type I, normally related great arteries; Type II, d-transposition of the great arteries; Type III, malpositions of the great arteries other than d-transposition; and Type IV, truncus arteriosus [6]. Some of the echocardiographic features are demonstrated in Figure 3.
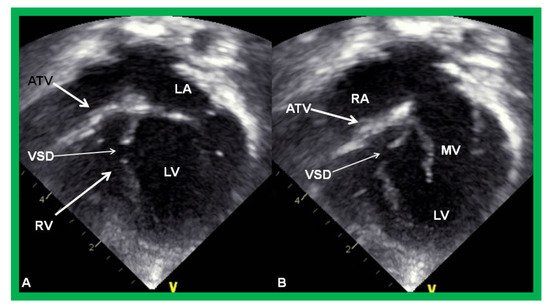
Figure 3. Echocardiograms in apical four-chamber views of an infant with tricuspid atresia demonstrating a dilated left ventricle (LV), a small right ventricle (RV), and a dense band of echoes at the site where the tricuspid valve echo should be (ATV; thick arrow). Images with closed (A) and open (B) mitral valve are shown; the tricuspid valve remains closed in both situations. A ventricular septal defect (VSD; thin arrow) is also shown. LA, Left atrium; RA, Right atrium.
2.3. Double-Inlet Left Ventricle
In DILV, the ventricle most frequently has LV morphology, although RV, mixed, indeterminate, or undifferentiated morphologies have been reported [1][15]. The main ventricle is largely a morphological LV with an outlet chamber connected to it which has a RV morphology. The AV valves may be normal (Figure 4), or one of them may be hypoplastic, stenotic, or even atretic. The great arteries are most frequently transposed and the aorta arises from the hypoplastic RV, and the pulmonary artery comes off the main LV chamber. L-transposition happens more often than d-transposition. The great vessels are normally related in 30% of cases. Double-outlet RV may be seen, in which both great vessels arise from the rudimentary RV. Pulmonary stenosis is present in two thirds of patients and such stenosis is seen irrespective of the great artery relationship. The stenosis may be at the valvar or subvalvar level, or the pulmonary valve/artery may be atretic. Subaortic obstruction may be present in patients with transposition of the great arteries and is due to the stenosis of the VSD or, more accurately, bulbo-ventricular foramen. Patients with subaortic obstruction often have associated aortic coarctation [1][15].
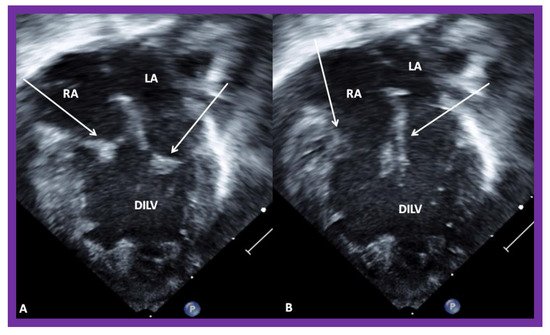
Figure 4. Echocardiograms in apical four-chamber views of a baby with double inlet left ventricle (DILV) with closed (A) and open (B) atrioventricular valves (arrows). The outlet chamber is not visualized in this view. LA, left atrium; RA, right atrium. Reproduced from Reference [15].
2.4. Unbalanced Atrioventricular Septal Defect
Defects in the atrial and ventricular septae, together with defects in one or both AV valves, described in the past as common AV canal or complete endocardial cushion defect [16], are now generally called AV septal defects (AVSDs). In the complete form, there is usually a large inlet VSD contiguous with a primum atrial septal defect (ASD), and a common AV valve with a single valve annulus. A variant of this, called the intermediate form, has the same features but with distinct and separate right and left AV valves. Transitional and partial forms are also described, and are similar to or the same as ostium primum ASDs. Complete AVSDs are also categorized based on the ventricular sizes, namely balanced and unbalanced (Figure 5) [17]. Unbalanced AVSDs comprise 10–15% of all complete AVSDs. The unbalanced types may be LV-dominant with a large LV and small RV, or RV-dominant with a large RV and small LV (Figure 5). RV-dominant AVSDs are more frequently seen [17].
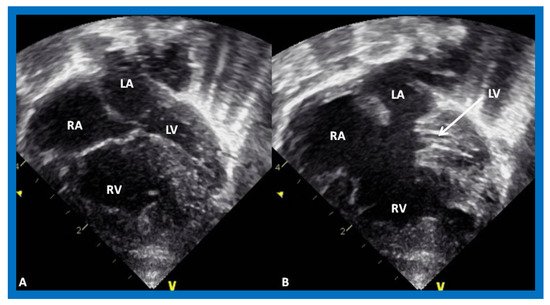
Figure 5. Echocardiograms in apical four-chamber views of an infant with complete atrioventricular septal defect with marked hypoplasia of the left ventricle (LV). LV in diastole (A) and in systole (B) are shown. LA, left atrium; RA, right atrium; RV, right ventricle.
2.5. Mitral Atresia with Normal Aortic Root
Mitral atresia may be seen with aortic atresia or a normal aortic root; the former is generally characterized as HLHS which was discussed in a preceding section. In this segment, mitral atresia with normal aortic root will be reviewed. While this division is arbitrary, it is considered appropriate [18] because of the differences in therapy for these two defects, in that HLHS babies almost always require the Norwood procedure [19][20] in the neonatal period, while babies with mitral atresia with normal aortic root need different types of intervention at different ages based on the associated cardiac defects [18]. Mitral atresia with normal aortic root is an uncommon complex CHD and is usually associated with several other defects. The atretic mitral valve may be an absent AV connection or an imperforate valve membrane. The left atrium (LA) is hypoplastic or small (Figure 6) and the right atrium is enlarged and hypertrophied. A PFO or ASD is generally seen; a PFO in 2/3rds of patients and an ASD in 1/3rd. A restrictive atrial septal defect may be present and, rarely, atrial septum is intact; in such cases the levoatriocardinal vein may help to decompress the LA. A single ventricle is most commonly associated ventricular anatomy, although in a few cases there are two ventricles. If two ventricles are present, the LV is small and communicates with the RV via a VSD which is usually small (Figure 6). The RV is always enlarged and hypertrophied. Transposition of the great vessels is frequently seen. Double-outlet right ventricle is present in some babies. In most patients the pulmonary valve is normal and not stenotic. However, in 25–30% of cases valvar and/or subvalvar stenosis or atresia is seen. The aortic valve and aortic root are near normal in size by definition. Coarctation of the aorta or interrupted aortic arch is seen in nearly 30% of cases. Such aortic obstruction is seen only in subjects with a normal pulmonary valve. PDA is seen in nearly 80% of patients [15][18].
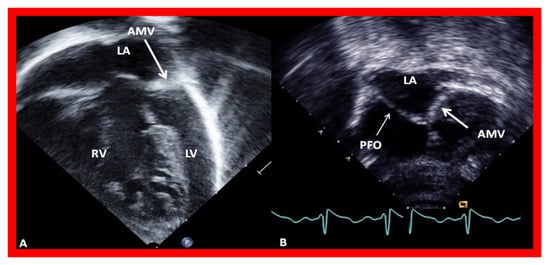
Figure 6. Echocardiograms in modified four-chamber views of two infants with mitral atresia, demonstrating atretic mitral valves (AMV), indicated by thick arrows. A small left atrium (LA) and left ventricle (LV) and a large right ventricle (RV) are also seen. The thin arrow in (B) shows a restrictive patent foramen ovale (PFO). All four chambers were shown in (A) while (B) focuses on the atria. Reproduced from Reference [15].
2.6. Other Heart Defects with Single Ventricle Physiology
There are other cardiac defects that may have functionally one ventricle. These will be briefed.
2.6.1. Pulmonary Atresia with Intact Ventricular Septum
Some children with pulmonary atresia with intact ventricular septum have severe hypoplasia of the RV, infudibular atresia or RV-dependant coronary circulation and become candidates for single ventricle repair [21][22][23][24]. Similarly, patients who end-up with very small RVs despite transcatheter and/or surgical procedures to improve the RV size will also join this group [21][22][23][24].
2.6.2. Ebstein’s Anomaly of the Tricuspid Valve
2.6.3. Heterotaxy Syndromes
3. Single Ventricle Physiology
In normal subjects the circulation is in series (Figure 7); the vena caval blood is emptied into the right atrium (RA), and from there is passed on to the RV, and pulmonary artery (PA) and is transported into the lungs for oxygenation. From there, it is returned to the LA, LV, and aorta and is passed on to the body for delivery of oxygen and nutrients. The resultant systemic arterial saturation is ≥96%. The blood is returned back to the vena cavae and the cycle is repeated (Figure 7).
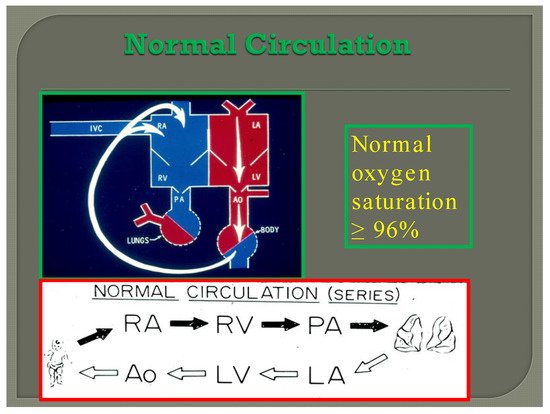
Figure 7. Diagram illustrating normal circulation in series (see the text) resulting systemic arterial oxygen saturation of ≥96%.
By contrast, in babies with single ventricle, the systemic (SBF) and pulmonary (PBF) blood flow returns mix with each other in the single functioning ventricle (Figure 8) with consequent lower systemic oxygen saturations [30]. The single ventricle then provides both the SBF and PBF (Figure 8). This admixture in the single ventricle causes systemic arterial oxygen desaturation. The O2 saturations vary between 75% and 85%, depending upon the quantity of PBF and pulmonary to systemic flow ratio (Qp:Qs).
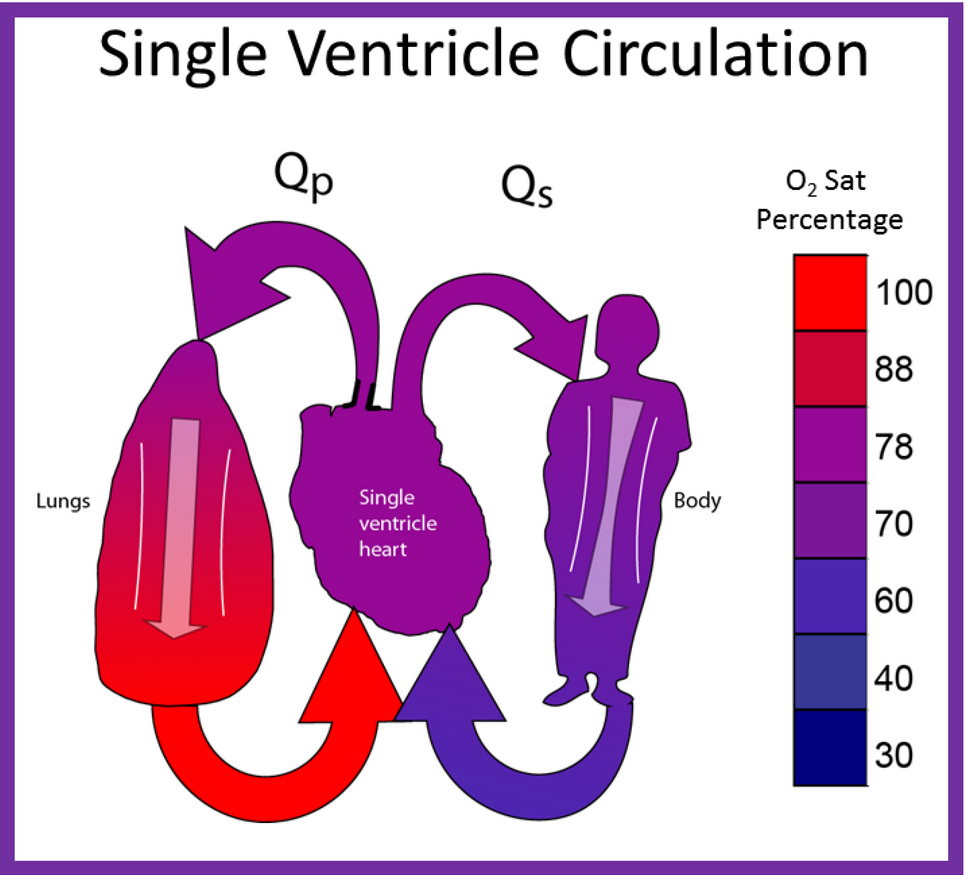
Figure 8. Diagrammatic portrayal of single ventricle circulation; the blood from both the lungs and body return to the single ventricle; this mixed blood is redistributed to the lungs (Qp) and body (Qs). Reproduced with permission from Reference [30].
The PBF is either derived from the pulmonary artery (PA) coming off of the single ventricle or from the level of the great vessels (patent ductus arteriosus, surgically placed aorta-pulmonary artery shunt (or RV to PA non-valved conduit), or aorto-pulmonary collateral vessels). The SBF is either unobstructed or obstructed.
Based on such a thought, these patients may be grouped into the following subsets [30]; this classification is helpful in determining the type of therapy that a given patient needs.
-
Group 1: Unobstructed Systemic Blood Flow
1A: Unobstructed SBF with unobstructed PBF
1B: Unobstructed SBF with moderately obstructed PBF
1C: Unobstructed SBF with severely obstructed (or atretic) PBF
-
Group 2: Obstructed Systemic Blood Flow
In summary, the systemic and pulmonary blood flow returns mix with each other with consequent lower systemic oxygen saturations (75–85%—Normal ≥96%). The systemic and pulmonary circuits work in-parallel rather than normal in-series circulation. Markedly increased PBF results in systemic hypo-perfusion and severely decreased PBF creates severe hypoxemia. Therefore, a delicate balance of the blood flows between the two circulations should be preserved.
4. Management Strategies to Address the Single Ventricle Defects
Since there is only one functioning ventricle, the overall objective is to allow the functioning ventricle (whether morphologically right or left) to supply the systemic circulation and to connect the systemic veins directly to the PAs (Fontan circulation—single ventricle repair). This procedure can’t be done in the neonate since PA pressure and resistance are high. Consequently, it is performed by staged procedures. The types of procedures and their timing have evolved over the years, as reviewed elsewhere [10][31][32][33][34], since their original description by Fontan, Kruetzer and their associates [35][36]. Currently, this is accomplished by staged total cavo-pulmonary connection (TCPC), devised by de Leval and associates [37]. This TCPC is undertaken in three stages: Stages I, II, and III.
4.1. Stage I—Management at the Time of Initial Presentation
The management at initial presentation, typically in the neonatal period, depends upon the pathophysiology of the defect itself plus the associated cardiac abnormalities, namely the status of PBF, the presence of obstruction to SBF, and other cardiac defects.
4.1.1. Status of Pulmonary Blood Flow
The PBF may be decreased, increased, or the PBF is adequate.
Reduced Pulmonary Blood Flow (Group 1C above)
In patients with reduced PBF, the ductus arteriosus should be kept patent by intravenous infusion of prostaglandin E1 (PGE1) at a dose of 0.05–0.1 mcg/kg/min. After the O2 saturation gets better, the dose of PGE1 is slowly, stepwise, decreased to 0.02–0.025 mcg/kg/min to lessen the adverse effects of the prostaglandins. Subsequent to achieving a stable baby and studies needed to firm up the diagnosis, a more stable way of supplying the PBF should be undertaken. Several methods of augmenting PBF have been utilized in the past, as reviewed elsewhere [38]. Of these, modified Blalock–Taussig (BT) shunt [39], ductal stenting [22][40][41], balloon valvuloplasty of the pulmonary valve (if the major obstruction is at the level of pulmonary valve) [42][43][44], and most recently connection of the RV outflow tract with the PA via a non-valved Gore-Tex tube [45] are currently available options. However, most surgeons use a modified BT shunt by placing of a Gore-Tex graft between the subclavian artery and the PA on the same side [39] (Figure 9) to address pulmonary oligemia.
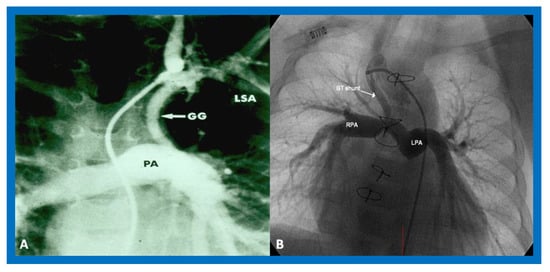
Figure 9. Cine-angiograms demonstrating patent Gore-Tex grafts (GG) following modified Blalock-Taussig (BT) shunt surgery in two patients. Note wide-open BT shunts with good visualization of the pulmonary artery (PA) in (A) of the right (RPA) and left (LPA) pulmonary arteries in (B).
Increased Pulmonary Blood Flow (Group 1A above)
A markedly elevated PBF produces congestive heart failure (CHF). These infants are initially stabilized with anticongestive therapy [46]. This should be followed by banding of the PA [47] (Figure 10), irrespective of control of CHF.
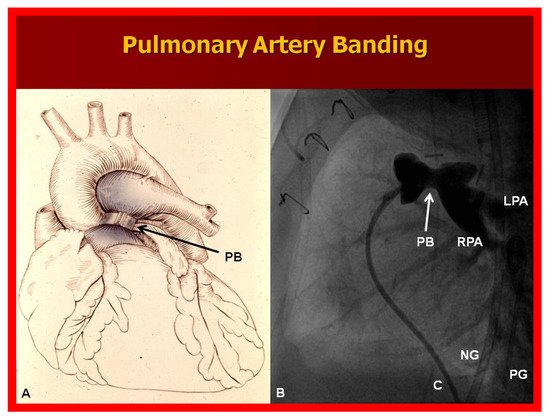
Figure 10. (A) Diagrammatic portrayal of pulmonary artery banding (PB) for infants with markedly elevated pulmonary blood flow and congestive heart failure. (B) Selected cine frame form pulmonary artery cineangiogram in straight lateral view demonstrating constriction of the pulmonary artery (PB; arrow) in an infant who had the banding procedure. C, catheter; LPA, left pulmonary artery; NG, nasogastric tube; PG, pigtail catheter; RPA, right pulmonary artery.
Adequate Pulmonary Blood Flow (Group 1B above)
Babies with a mildly increased or near normal PBF with O2 saturations in the low 80s do not exhibit significant symptoms and, are less cyanotic than babies with pulmonary oligemia and do not require any treatment and may be followed until Stage II.
4.1.2. Obstructed Systemic Blood Flow (Group 2 above)
The majority of these babies belong to HLHS syndrome or its variants and require Norwood operation [19][20] with either Modified BT [39] or Sano [48][49] shunt (Figure 11). In some institutions, cardiac transplantation [50] is employed to address this lesion.
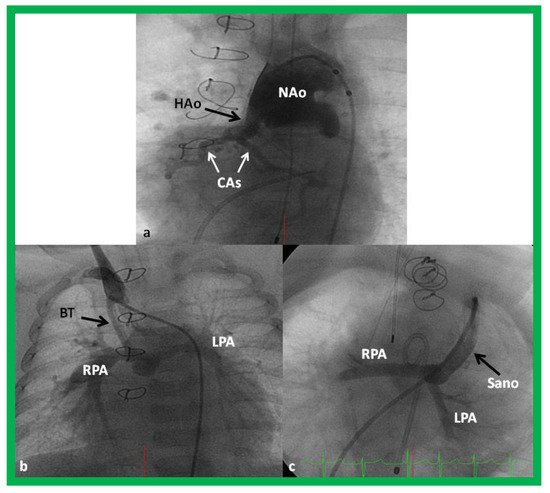
Figure 11. Cineangiogrames demonstrating the Norwood procedure, showing the neoaorta (NAo) and hypoplastic old aorta (HAo). The latter supplies the coronary arteries (CAs) as shown in (a). A Blalock-Taussig (BT) shunt in seen in (b) and a Sano shunt in (c) from two other babies are also shown. This is Stage I of the Fontan procedure for infants with hypoplastic left heart syndrome. LPA, left pulmonary artery; RPA, right pulmonary artery. Reproduced from Reference [33].
4.1.3. Other Cardiac Abnormalities
Some of the babies may have other cardiac abnormalities such as intra-cardiac obstruction, aortic arch obstruction, or atrioventricular valve insufficiency at presentation or may develop such abnormalities during the succeeding period. These will be reviewed.
Intra-Cardiac Obstruction
Intra-cardiac obstruction may take place either at the atrial or at the ventricular level.
Inter-Atrial Obstruction
The total systemic or pulmonary venous return must pass through the PFO in babies with atretic atrioventricular valves and/or atretic semi-lunar valves. These defects are listed in Table 1.
Table 1. Inter-atrial Obstruction.
| 1 | Pulmonary atresia |
| 2 | Tricuspid atresia |
| 3 | Mitral atresia |
| 4 | Hypoplastic left heart syndrome |
There is a tendency for the PFO to remain open because of a persistence of fetal flow patterns. However, sometimes the PFO becomes obstructed. Evidence for obstructed PFO may be present either with systemic or pulmonary venous congestion and is confirmed by a small-sized PFO by 2-dimentional echo and high velocity flow across it by Doppler studies. The PFO may be enlarged by balloon atrial septostomy (Figure 12) [51]. Such a procedure is usually successful, especially in the early neonatal period, because the septum primum (lower margin of the PFO) is thin and flail and can be ruptured by balloon septostomy.
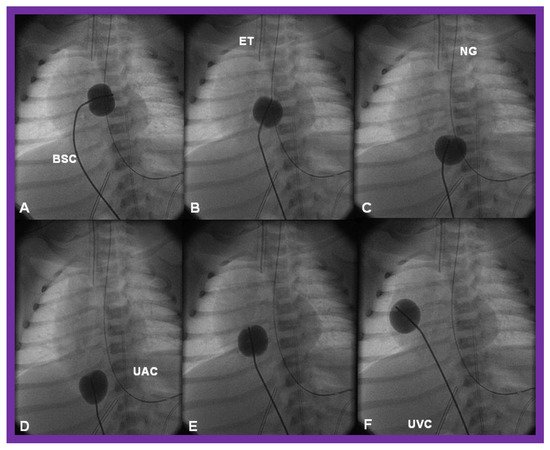
Figure 12. Cinfluroscopic frames demonstrating the procedure of Rashkind’s balloon septostomy. Initially the balloon is inflated in the left atrium (A). The balloon septostomy catheter (BSC) is rapidly and forcefully pulled into the right atrium (B) and inferior vena cava (C,D) and quickly advanced back into the right atrium (E,F). The entire procedure is done as one single motion. Rapid advancement of the BSC into the right atrium (E,F) is done in order to avoid inadvertent occlusion of the inferior vena cava if failure to deflate the balloon occurs (this is quite rare). At about the same time the balloon is deflated. ET, endotracheal tube; NG, nasogastric tube; UAC, umbilical arterial catheter; UVC, umbilical venous catheter.
When PFO obstruction develops during the later part of the neonatal period or in older infants, balloon atrial septostomy is unlikely to be successful, and in such situations, alternative methods such as blade atrial septostomy [52], static balloon dilatation [53][54], or stent placement [55][56][57] may have to be used to accomplish the relief of inter-atrial obstruction. If transcatheter methods are not feasible or not successful, surgical atrial septectomy is necessary. If there is no PFO and the atrial septum is intact, the atrial septum may be perforated either by Brockenbrough technique [58] or radiofrequency perforation [59]. This should be followed by static dilatation [53][54] or stent placement across the atrial septum [55][56][57]. For a detailed discussion of these transcatheter techniques, the interested reader may review the author’s previous publications [57][60].
In babies with single ventricle and mitral atresia, inter-atrial obstruction is frequently present [61][62]. In such infants, predictable fall in pulmonary vascular resistance (PVR) takes place after relief of atrial obstruction either by balloon septostomy or surgery [62]. Therefore, banding of the PA should be performed without hesitation at the time of alleviating the atrial septal obstruction in order to reduce the probability for CHF, reduce the PVR and PA pressure, prevent pulmonary vascular obstructive disease (PVOD), and pave the way for Fontan circulation [61][62].
Inter-Ventricular Obstruction
In some complex cardiac defects, namely, tricuspid atresia, double-inlet left ventricle, and double-outlet right (or left) ventricle, the VSD or bulbo-ventricular foramen, as the case may be, is very small and obstructive at presentation in the neonatal period or spontaneously becomes smaller with time [13][14][63][64]. Such a reduction in the size of the VSD or bulbo-ventricular foramen causes sub-pulmonary obstruction, producing reduced pulmonary blood flow or subaortic narrowing, resulting in obstruction to systemic blood flow (Table 2).
Table 2. Inter-Ventricular Obstruction.
| 1 | Tricuspid atresia |
| 2 | Double-inlet left ventricle |
| 3 | Double-outlet right (or left) ventricle |
If such a closure produces diminished pulmonary blood flow, the management is similar to that discussed in the “Decreased Pulmonary Blood Flow” section above (PGE1 and modified BT shunt). If the VSD/bulbo-ventricular foramen closure results in obstruction to SBF, the narrowing is either relieved directly by enlarging the VSD or the obstruction is bypassed by the Damus–Kaye–Stansel (DKS) procedure (anastomosis of the divided pulmonary artery to the ascending aorta either directly or via a prosthetic conduct) (Figure 13) [14][65]. Most commonly DKS is performed instead of directly enlarging the VSD. However, it should be noted that the development of a significant inter-ventricular obstruction that requires intervention in the neonate is infrequent.
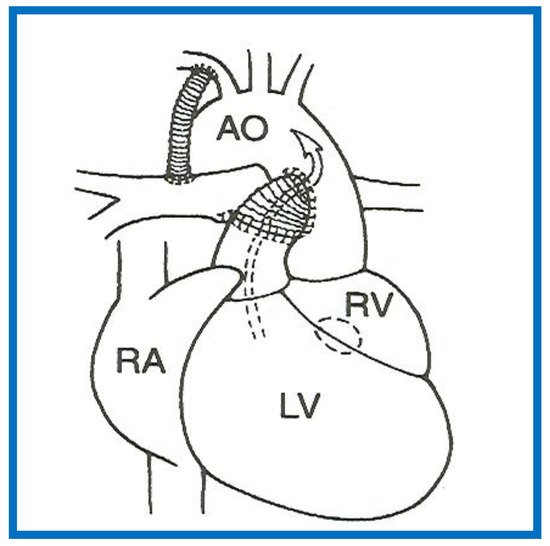
Figure 13. A Line drawing illustrating Damus-Kaye-Stansel procedure. The left ventricular (LV) blood flows via the ventricular septal defect (circle) and right ventricle (RV) into the aorta (Ao). If the VSD is small and restrictive, causing “subaortic” obstruction, this obstruction may be circumvented by connecting the proximal stump of the divided pulmonary artery to the Ao directly or a via a non-valved conduit. The pulmonary arteries are supplied with a Blalock-Taussig shunt. Concept derived from References [13][14].
Aortic Obstruction
Aortic arch obstruction may occur in the form of aortic coarctation or interrupted aortic arch. Both will be briefly reviewed.
Aortic Coarctation
Aortic coarctation is a descending aortic abnormality with constriction of the aortic arch distal to the left subclavian artery at about the level of ductal insertion [66]. In the neonate, it is frequently associated with other defects such as PDA, VSD and other complex CHDs such as tricuspid atresia with transposition of the great arteries and DILV with transposition of the great arteries. Initial medical management by infusion of PGE1 to bypass the coarctation and anti-congestive treatment, if CHF is present, is suggested. In most institutions the neonatal coarctations are addressed by surgery. Although the immediate success rate following balloon angioplasty is good [67][68][69], because of a high recurrence rate [69][70][71] associated with balloon therapy in the neonate, surgery is preferred. However, in special circumstances when surgical risk is high or contraindicated, balloon dilatation may be used as an initial treatment option [67][68][69][72].
Interrupted Aortic Arch
In aortic arch interruption, there is a total loss of luminal continuity between the arch of the aorta and the descending aorta [73] and the condition is classified into types A, B, and C [74] depending upon the site of interruption. The initial management is by PGE1 infusion to allow the ductus to open and restore systemic perfusion. Then, end-to-end anastomosis after removal of the ductal tissue is performed. In cases where the aortic arch can’t be mobilized, an interposition Gore-Tex graft is inserted to bridge the gap. In any baby with interrupted aortic arch, especially in association with single ventricle physiology, arch repair should be promptly performed.
Atrio-Ventricular Valve Insufficiency
Mild AV valve insufficiency does not require any treatment. However, moderate to severe AV valve insufficiency (tricuspid, mitral or common AV valve) should be addressed promptly. Initially medical management with angiotensin-converting-enzyme (ACE) inhibitors (Captopril/Enalopril) may be used. If no adequate improvement with medical therapy is seen, surgical valvuloplasty or valve replacement may become necessary.
4.2. Stage II—Bidirectional Glenn
Independent of the nature of palliative intervention during the neonatal period, bidirectional Glenn operation [75] by anastomosis of the superior vena cava (SVC) to the right PA, end-to-side (Figure 14) is undertaken at an approximate age of six months. If a prior BT or Sano shunt is present, it is ligated at the time of bidirectional Glenn. While performing the procedure at six months is commonly accepted, the Glenn can be performed as early as three months subject to demonstrating normal PA pressures and anatomy.
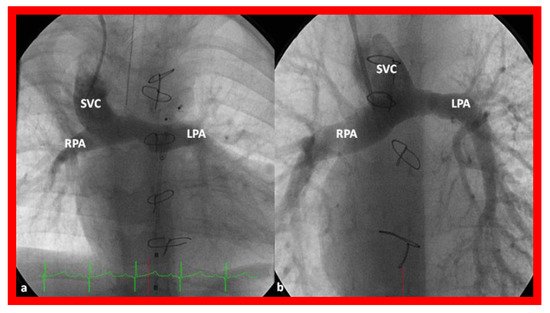
Figure 14. Cineangiographic frames illustrating bidirectional Glenn procedure, i.e., anastomosis of the superior vena cava (SVC) to the right pulmonary artery (RPA)] in two different children is shown in (a,b) (Stage II). Unobstructed blood flow from the SVC to the right (RPA) and left (LPA) pulmonary arteries is seen. Reproduced from Reference [33].
In babies who have persistent left SVC, bilateral bidirectional Glenn (Figure 15) is performed particularly in patients with a small or absent left innominate vein. A bidirectional Glenn procedure may also be undertaken for patients with infrahepatic interruption of the IVC with azygos or hemiazygos continuation, and such a procedure may be called the Kawashima procedure.
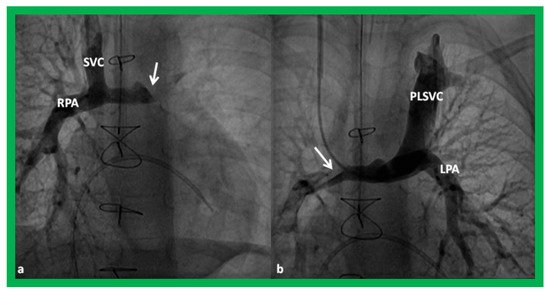
Figure 15. Cineangiographic frames demonstrating a bilateral bidirectional Glenn procedure (Stage II). In (a), angiogram of the superior vena cava (SVC) illustrates opacification of the right pulmonary artery (RPA). The arrow in (a) shows the unopacified blood from a persistent left superior vena cava (PLSVC). In (b), an injection into the PLSVC illustrates opacification of the left pulmonary artery (LPA). The arrow in (b) shows the unopacified blood from the right SVC. Unobstructed flow from the respective SVCs into the pulmonary arteries is clearly seen. Reproduced from Reference [33].
Prior to undertaking the bidirectional Glenn procedure, it must be ensured that the PA pressures are normal and the branch PAs are adequate in size. This is most often accomplished by cardiac catheterization and cineangiography. However, some institutions employ echocardiogrms or other imaging studies such as magnetic resonance imaging (MRI) or computed tomography (CT) to accomplish this goal. If stenosis of the PAs is present, it may be relieved with balloon angioplasty or stent placement, as appropriate, or it may be repaired while performing the bidirectional Glenn procedure. If atrioventricular valve regurgitation, aortic coarctation, subaortic obstruction, and other abnormalities are present, they should also be repaired at the time of bidirectional Glenn surgery.
4.3. Stage III—Fontan/Kruetzer Procedure
Although the author prefers the term “Fontan/Kruetzer Procedure” because of the simultaneous description of the procedure by both groups [35][36], it is more commonly referred to as “Fontan Procedure” in the literature and therefore, it will be so used in the rest of the presentation. During the final stage, the IVC blood flow is rerouted into the PA. At the same time a fenestration between the conduit and the atrial mass is created. Arbitrary division of these procedures into Stage IIIA (diversion of IVC into the PA) and Stage IIIB (closure of the fenestration) [33][34] may be undertaken.
4.3.1. Stage IIIA
In the Stage IIIA, the TCPC may be accomplished by redirecting the IVC blood flow into the PA either by a lateral tunnel [76][77] or by an extra-cardiac, non-valved conduit (Figure 16) [78][79]. This surgery is typically carried out anytime from one to two years of age, frequently one year after the bidirectional Glenn. At the present time, the majority of surgeons favor an extra-cardiac conduit to achieve the final stage of Fontan. It also appears that most surgeons create a fenestration, 4–6 mm in size, between the conduit and the atria (Figure 16) [80]. Whereas creation of fenestration during the Fontan surgery was originally suggested for patients with high-risk [80][81], most surgeons and pediatric intensivists appear to opt for fenestration, because the creation of fenestration during the Fontan decreases mortality rate and lessens the morbidity during the immediate postoperative period [33].
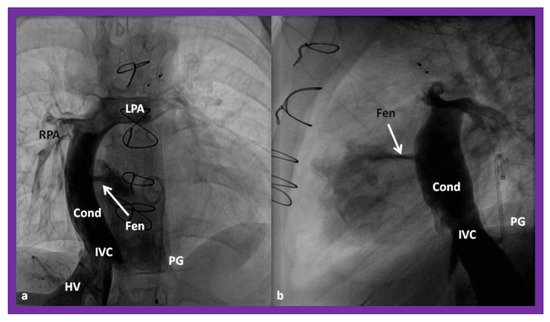
Figure 16. Cineangiographic frames in postero-anterior (a) and lateral (b) projections, illustrating Stage IIIA of the Fontan procedure in which the inferior vena caval (IVC) blood flow is diverted into the pulmonary arteries via a non-valve conduit (Cond). The flow via the fenestration (Fen) is shown by the arrows in a and b. HV, hepatic veins; LPA, left pulmonary artery; PG, pigtail catheter in the descending aorta; RPA, right pulmonary artery. Modified from Reference [33].
4.3.2. Stage IIIB
During the Stage IIIB, the fenestration is occluded (Figure 17) by transcatheter methods [33][80][82][83][84], usually 6–12 months following Stage IIIA Fontan. In the past, all previously available ASD occluding devices [80][82][83][84] were used for fenestration closure. However, at the present time, Amplatzer Septal Occluders are the most regularly used devices to accomplish fenestration closures. Any other residual shunts may also be addressed by device closure.
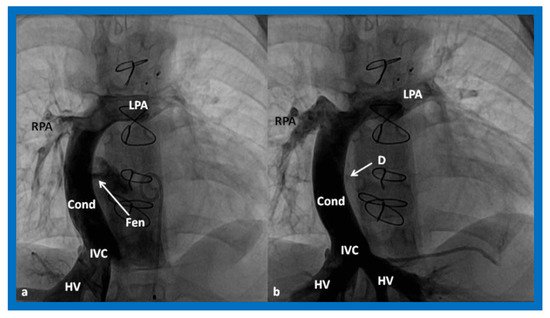
Figure 17. (a). Cineangiogram in antero-posterior view, illustrating Stage IIIA of the Fontan operation, diverting the inferior vena caval (IVC) blood flow into the pulmonary arteries via a non-valve conduit (Cond). Fenestration (Fen) is shown by the arrow in (a). The Fen is occluded with an Amplatzer device (D), shown by the arrow in (b) (Stage IIIB). HV, hepatic veins; LPA, left pulmonary artery; RPA, right pulmonary artery. Reproduced from Reference [33].
References
- Rao, P.S. Diagnosis and management of cyanotic congenital heart disease: Part II. Indian. J. Pediatr. 2009, 76, 297–308.
- Noonan, J.A.; Nadas, A.S. The hypoplastic left heart syndrome. Pediat. Clin. N. Am 1958, 5, 1029–1056.
- Rao, P.S.; Striepe, V.; Kambam, J. Hypoplastic left heart syndrome. In Cardiac Anesthesia for Infants and Children; Kambam, J., Ed.; Mosby-Year Book: St. Louis, MO, USA, 1994; pp. 296–309.
- Alapati, S.; Rao, P.S. Hypoplastic Left Heart Syndrome in the Neonate. Neonatol. Today 2011, 6, 1–9.
- Rao, P.S.; Alapati, S. Hypoplastic left heart syndrome. In A Comprehensive Approach to Management of Congenital Heart Diseases; Vijayalakshmi, I.B., Rao, P.S., Chugh, R., Eds.; Jaypee Publications: New Delhi, India, 2013; pp. 662–678.
- Rao, P.S. A unified classification for tricuspid atresia. Am. Heart J. 1980, 99, 799–804.
- Rao, P.S. (Ed.) Terminology: Tricuspid atresia or univentricular heart? In Tricuspid Atresia; Futura Publishing Co.: Mount Kisco, NY, USA, 1982; pp. 3–6.
- Rao, P.S. (Ed.) Tricuspid Atresia; Futura Publishing Co.: Mount Kisco, NY, USA, 1982.
- Rao, P.S. (Ed.) Tricuspid Atresia, 2nd ed.; Futura Publishing Co.: Mount Kisco, NY, USA, 1992.
- Rao, P.S. Pediatric Tricuspid Atresia. Medscape Drugs & Diseases. Available online: (accessed on 17 January 2017).
- Rao, P.S. Tricuspid atresia. In Pediatric Cardiovascular Medicine, 2nd ed.; Moller, J.H., Hoffman, J.I.E., Eds.; Wiley-Blackwell/A John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2012; pp. 487–508.
- Rao, P.S. Tricuspid atresia. In A Comprehensive Approach to Management of Congenital Heart Diseases; Vijayalakshmi, I.B., Rao, P.S., Chugh, R., Eds.; Jaypee Publications: New Delhi, India, 2013; pp. 397–413.
- Rao, P.S. Natural history of the ventricular septal defect in tricuspid atresia and its surgical implications. Br Heart J. 1977, 39, 276–288.
- Rao, P.S. Further observations on the spontaneous closure of physiologically advantageous ventricular septal defects in tricuspid atresia: Surgical implications. Ann. Thorac. Surg. 1983, 35, 121–131.
- Rao, P.S. Other cyanotic heart defects in the neonate. In Perinatal Cardiology: A Multidisciplinary Approach; Rao, P.S., Vidyasagar, D., Eds.; Cardiotext Publishing: Minneapolis, MN, USA, 2015.
- Titus, J.L.; Rastelli, G.C. Anatomic features of persistent common atrioventricular canal. In Artioventricular Canal Defects; Feldt, R.H., Ed.; WB Saunders Company: Philadelphia, PA, USA, 1976; pp. 13–35.
- Rao, P.S.; Harris, A.D. Recent advances in managing septal defects: Ventricular septal defects and atrioventricular septal defects. F1000 Res. 2018, 7.
- Balaguru, D.; Rao, P.S. Mitral Atresia (With Normal Aortic Root). In A Comprehensive Approach to Management of Congenital Heart Diseases; Vijayalakshmi, I.B., Rao, P.S., Chugh, R., Eds.; Jaypee Publications: New Delhi, India, 2013; pp. 458–467.
- Norwood, W.I.; Kirklin, J.K.; Sanders, S.P. Hypoplastic left heart syndrome: Experience with palliative surgery. Am. J. Cardiol. 1980, 45, 87–91.
- Norwood, W.I.; Lang, P.; Hansen, D.D. Physiologic repair of aortic atresia-hypoplastic left heart syndrome. N. Engl. J. Med. 1983, 308, 23–26.
- Rao, P.S. Comprehensive management of pulmonary atresia with intact ventricular septum. Ann. Thorac. Surg. 1985, 40, 409–413.
- Siblini, G.; Rao, P.S.; Singh, G.K.; Tinker, K.; Balfour, I.C. Transcatheter management of neonates with pulmonary atresia and intact ventricular septum. Cathet. Cardiovasc. Diagn. 1997, 42, 395–402.
- Rao, P.S. Pulmonary atresia with intact ventricular septum. Curr. Treat. Options Cardiovasc. Med. 2002, 4, 321–336.
- Balaguru, D.; Rao, P.S. Pulmonary Atresia with Intact Ventricular Septum. In Perinatal Cardiology: A Multidisciplinary Approach; Rao, P.S., Vidyasagar, D., Eds.; Cardiotext Publishing: Minneapolis, MN, USA, 2015.
- Rao, P.S. Tricuspid valve abnormalities other than tricuspid atresia. In Fetal and Neonatal Cardiology; Long, W.A., Ed.; W.B. Saunders: Philadelphia, PA, USA, 1990; pp. 541–550.
- Balaguru, D.; Rao, P.S. Ebstein’s anomaly of the tricuspid valve. In Perinatal Cardiology: A Multidisciplinary Approach; Rao, P.S., Vidyasagar, D., Eds.; Cardiotext Publishing: Minneapolis, MN, USA, 2015.
- Balaguru, D.; Rao, P.S. Diseases of the tricuspid valve (Ebstein’s anomaly, tricuspid stenosis and regurgitation). In A Comprehensive Approach to Management of Congenital Heart Diseases, 2nd ed.; Vijayalakshmi, I.B., Rao, P.S., Chugh, R., Eds.; Jaypee Publications: New Delhi, India, 2019; Chapter 29.
- Rao, P.S. Cardiac Malpositions including Heterotaxy Syndromes. In Perinatal Cardiology: A Multidisciplinary Approach; Rao, P.S., Vidyasagar, D., Eds.; Cardiotext Publishing: Minneapolis, MN, USA, 2015; Chapter 36.
- Rao, P.S. Cardiac Malposition. In PG Textbook of Pediatrics, 2nd ed.; Gupta, P., Menon, P.S.N., Ramji, S., Lodha, R., Eds.; Jaypee Brothers Medical Publishers (P) Ltd.: New Delhi, India, 2018.
- Yarrabolu, T.; Douglas, W.I. Single ventricle physiology. In Perinatal Cardiology: A Multidisciplinary Approach; Rao, P.S., Vidyasagar, D., Eds.; Cardiotext Publishing: Minneapolis, MN, USA, 2015.
- Chopra, P.S.; Rao, P.S. Corrective surgery for tricuspid atresia: Which modifications of Fontan-Kreutzer procedure should be used? A review. Am. Heart. J. 1992, 123, 758–767.
- Rao, P.S.; Chopra, P.S. Modification of Fontan-Kreutzer procedure for tricuspid atresia: Can a choice be made? In Tricuspid Atresia, 2nd ed.; Rao, P.S., Ed.; Futura Publishing Co.: Mount Kisco, NY, USA, 1992; pp. 361–375.
- Rao, P.S. Fontan operation: Indications, short and long term outcomes. Indian J. Pediatr. 2015, 82, 1147–1156.
- Rao, P.S. Fontan operation: A comprehensive review. In Fontan Surgery; Khan, I., Ed.; InTechOpen: Rijeka, Croatia, 2020.
- Fontan, F.; Baudet, E. Surgical repair of tricuspid atresia. Thorax 1971, 26, 240–248.
- Kreutzer, G.; Bono, H.; Galindez, E. Una operacion para la correccion de la atresia tricuspidea. In Proceedings of the Ninth Argentinean Congress of Cardiology, Buenos Aires, Argentina, 31 October–6 November 1971.
- De Leval, M.R.; Kilner, P.; Gewillig, M.; Bull, C.; McGoon, D.C. Total cavopulmonary connection: A logical alternative to atriopulmonary connection for complex Fontan operation. J. Thorac. Cardiovasc. Surg. 1988, 96, 682–695.
- Rao, P.S.; Covitz, W.; Chopra, P.S. Principles of palliative management of patients with tricuspid atresia. In Tricuspid Atresia, 2nd ed.; Rao, P.S., Ed.; Futura Publishing Co.: Mt. Kisco, NY, USA, 1992; pp. 297–320.
- De Leval, M.; McKay, R.; Jones, M.; Stark, J.; McCartney, F.J. Modified Blalock-Taussig shunt: Use of subclavian orifice as a flow regulator in prosthetic systemic-pulmonary artery shunts. J. Thorac. Cardiovasc. Surg. 1981, 18, 112–119.
- Gibbs, J.L.; Orhan, U.; Blackburn, M.E.C.; Wren, C.; Hamilton, J.R.; Watterson, K.G. Fate of stented arterial duct. Circulation 1999, 99, 2621–2625.
- Alwi, M.; Choo, K.K.; Latiff, H.A.; Kandavello, G.; Hasri Samion, H.; Mulyadi, M.D. Initial results and medium-term follow-up of stent implantation of patent ductus arteriosus in duct-dependent pulmonary circulation. J. Amer. Coll. Cardiol. 2004, 44, 438–445.
- Rao, P.S.; Brais, M. Balloon pulmonary valvuloplasty for congenital cyanotic heart defects. Amer. Heart. J. 1988, 115, 1105–1110.
- Rao, P.S.; Wilson, A.D.; Thapar, M.K.; Brais, M. Balloon pulmonary valvuloplasty in the management of cyanotic congenital heart defects. Cathet. Cardiovasc. Diagn. 1992, 25, 16–24.
- Rao, P.S. Pulmonary valve disease: Pulmonary valve in cyanotic heart defects with pulmonary oligemia. In Interventions in Structural, Valvular and Congenital Heart Disease; Sievert, H., Qureshi, S.A., Wilson, N., Hijazi, Z., Eds.; CRC Press: Boca Raton, FL, USA, 2014; pp. 297–308.
- Alapati, S.; Rao, P.S. Tetralogy of Fallot. In A Multidisciplinary Approach to Perinatal Cardiology; Rao, P.S., Vidyasagar, D., Eds.; Cambridge Scholars Publishing: New Castle Upon Tyne, UK, 2021; Volume 2, pp. 203–246.
- Rao, P.S. Congenital heart disease. In Conn’s Current Therapy; Rakel, R.E., Ed.; WB Saunders Co: Philadelphia, PA, USA, 1989; pp. 201–213.
- Muller, W.H., Jr.; Danimann, J.F., Jr. The treatment of certain congenital malformations of the heart by the creation of pulmonic stenosis to reduce pulmonary hypertension and excessive pulmonary blood flow; a preliminary report. Surg. Gynec. Obst. 1952, 95, 213–219.
- Sano, S.; Ishino, K.; Kawada, M.; Arai, S.; Kasahara, S.; Asai, T.; Masuda, Z.; Takeuchi, M.; Ohtsuki, S. Right ventricle-pulmonary artery shunt in first-stage palliation of hypoplastic left heart syndrome. J. Thorac. Cardiovasc. Surg. 2003, 126, 504–509.
- Sano, S.; Ishino, K.; Kado, H.; Shiokawa, Y.; Sakamoto, K.; Yokota, M.; Kawada, M. Outcome of right ventricle-to-pulmonary artery shunt in first-stage palliation of hypoplastic left heart syndrome: A multi-institutional study. Ann. Thorac. Surg. 2004, 78, 1951–1957, discussion 1957–1958.
- Bailey, L.; Concepcion, W.; Shattuk, H.; Huang, L. Method of heart transplantation for treatment of hypoplastic left heart syndrome. J. Thorac. Cardiovasc. Surg. 1986, 92, 1–9.
- Rashkind, W.J.; Miller, W.W. Creation of an atrial septal defect without thoracotomy. J. Am. Med. Assoc. 1966, 196, 991–992.
- Park, S.C.; Neches, W.H.; Zuberbuhler, J.R.; Lenox, C.C.; Mathews, R.A.; Fricker, F.J.; Zoltun, R.A. Clinical use of blade septostomy. Circulation 1978, 58, 600–606.
- Shrivatsava, S.; Radhakrishnan, S.; Dev, V.; Singh, L.S.; Rajani, M. Balloon dilatation of atrial septum in complete transposition of great arteries—A new technique. Indian Heart J. 1987, 39, 298–300.
- Rao, P.S. Static balloon dilation of atrial septum (Editorial). Am. Heart. J. 1993, 125, 1826–1827.
- Gewillig, D.; Boshoff, L.; Mertens, L. Creation with a stent of an unrestrictive lasting atrial communication. Cardiol. Young 2002, 12, 404–407.
- Eicken, A.; Gildein, H.P.; Schreiber, C.; Balling, G.; Hess, J. Stenting of a restrictive foramen ovale in a patient with hypoplastic left heart syndrome. Intern. J. Cardiol. 2006, 113, 254–256.
- Rao, P.S. Neonatal Catheter Interventions. In Cardiac Catheterization and Imaging (From Pediatrics to Geriatrics); Vijayalakshmi, I.B., Ed.; Jaypee Publications: New Delhi, India, 2015; pp. 388–432.
- Atz, A.M.; Feinstein, J.A.; Jonas, R.A.; Perry, S.B.; Wessel, D.L. Preoperative management of pulmonary venous hypertension in hypoplastic left heart syndrome with restrictive atrial septal defect. Am. J. Cardiol. 1999, 83, 1224–1228.
- Justino, H.; Benson, L.N.; Nykanen, D.G. Transcatheter creation of an atrial septal defect using radiofrequency perforation. Catheter. Cardiovasc. Interv. 2001, 54, 83–87.
- Rao, P.S. Catheter interventions in the neonate. In A Multidisciplinary Approach to Perinatal Cardiology; Rao, P.S., Vidyasagar, D., Eds.; Cambridge Scholars Publishing: New Castle Upon Tyne, UK, 2021; Volume 1, pp. 646–742.
- Rao, P.S.; Moore, H.V.; Strong, W.B. Surgery for mitral atresia and interatrial obstruction without pulmonic stenosis (Letter). Circulation 1980, 62, 201–202.
- Rao, P.S.; Kulangara, R.J.; Moore, H.V.; Strong, W.B. Syndrome of single ventricle without pulmonic stenosis but with left atrioventricular valve atresia and interatrial obstruction: Palliative management with simultaneous atrial septostomy and pulmonary artery banding. J. Thorac. Cardiovasc. Surg. 1981, 81, 127–130.
- Rao, P.S. Physiologically advantageous ventricular septal defects (Letter). Pediatr. Cardiol. 1983, 4, 59–62.
- Rao, P.S. Subaortic obstruction after pulmonary artery banding in patients with tricuspid atresia and double-inlet left ventricle and ventriculoarterial discordance (Letter). J. Am. Coll. Cardiol. 1991, 18, 1885–1886.
- Stansel, H.C., Jr. A new operation for d- transposition of the great vessels. Ann. Thorac. Surg. 1975, 19, 565–567.
- Rao, P.S. Coarctation of the aorta. In Secondary Forms of Hypertension; Seminars in Nephrology; Kurtzman, N.A., Ram, C.V.S., Eds.; W.B. Saunders: Philadelphia, PA, USA, 1995; Volume 15, pp. 81–105.
- Rao, P.S.; Thapar, M.K.; Galal, O.; Wilson, A.D. Follow-up results of balloon angioplasty of native coarctation in neonates and infants. Am. Heart. J. 1990, 120, 1310–1319.
- Rao, P.S. Current status of balloon angioplasty for neonatal and infant aortic coarctation. Prog. Pediat. Cardiol. 2001, 14, 35–44.
- Rao, P.S.; Jureidini, S.B.; Balfour, I.C.; Singh, G.K.; Chen, S.C. Severe aortic coarctation in infants less than 3 months: Successful palliation by balloon angioplasty. J. Interv. Cardiol. 2003, 15, 203–208.
- Reddington, A.N.; Booth, P.; Shore, D.F.; Rigby, M.L. Primary balloon dilatation of coarctation in neonates. Br Heart J. 1990, 64, 277–281.
- Rao, P.S.; Galal, O.; Smith, P.A.; Wilson, A.D. Five-to-nine-year follow-up results of balloon angioplasty of native aortic coarctation in infants and children. J. Am. Coll. Cardiol. 1996, 27, 462–470.
- Rao, P.S. Should balloon angioplasty be used as a treatment of choice for native aortic coarctations? J. Invasive Cardiol. 1996, 8, 301–308.
- Sharma, S.K.; Rao, P.S. Interrupted aortic arch. In The Encyclopedic Reference of Molecular Mechanisms of Disease; Springer: Berlin/Heidelberg, Germany; New York, NY, USA; Tokyo, Japan, 2008.
- Celoria, G.C.; Patton, R.B. Congenital absence of the aortic arch. Am. Heart. J. 1959, 58, 407–413.
- Hopkins, R.A.; Armstrong, S.E.; Serwer, G.A.; Peterson, R.J.; Oldham, H.N., Jr. Physiologic rationale for a bidirectional cavopulmonary shunt: A versatile complement to the Fontan principle. J. Thorac. Cardiovasc. Surg. 1985, 90, 391–398.
- Puga, F.J.; Chiavarelli, M.; Hagler, D.J. Modifications of the Fontan operation applicable to patients with left atrioventricular valve atresia or single atrioventricular valve. Circulation 1987, 76 (Suppl. III), III53–III60.
- Jonas, R.A.; Castaneda, A.R. Modified Fontan procedure: Atrial baffle and systemic venous to pulmonary artery anastomotic techniques. J. Cardiac Surg. 1988, 3, 91–96.
- Marcelletti, C.; Corno, A.; Giannico, S.; Marino, B. Inferior vena cava-pulmonary artery extra-cardiac conduit. A new form of right heart bypass. J. Thorac. Cardiovasc. Surg. 1990, 100, 228–232.
- Marcelletti, C.; Iorio, F.S.; Abella, R.F. Late results of extra-cardiac Fontan repair. Semin. Thorac. Cardiovasc. Surg. Pediatr. Card. Surg. Annu. 1999, 2, 131–141.
- Bridges, N.D.; Lock, J.E.; Castaneda, A.R. Baffle fenestration with subsequent transcatheter closure. Modification of the Fontan operation for patients at increased risk. Circulation 1990, 82, 1681–1689.
- Laks, H.; Pearl, J.M.; Haas, G.S.; Drinkwater, D.C.; Milgalter, E.; Jarmakani, J.M.; Isabel-Jones, J.; George, B.L.; Williams, R.G. Partial Fontan: Advantages of an adjustable interatrial communication. Ann. Thorac. Surg. 1991, 52, 1084–1094, discussion 1094–1095.
- Rao, P.S.; Chandar, J.S.; Sideris, E.B. Role of inverted buttoned device in transcatheter occlusion of atrial septal defects or patent foramen ovale with right-to-left shunting associated with previously operated complex congenital cardiac anomalies. Am. J. Cardiol. 1997, 80, 914–921.
- Goff, D.A.; Blume, E.D.; Gauvreau, K.; Mayer, J.E.; Lock, J.E.; Jenkins, K.J. Clinical Advances in Complex Valvular Disease 24 outcome of fenestrated Fontan patients after closure: The first 10 years. Circulation 2000, 102, 2094–2099.
- Boudjemline, Y.; Bonnet, D.; Sidi, D.; Agnoletti, G. Closure of extracardiac Fontan fenestration by using the Amplatzer duct occluder. Arch. des Mal. du Coeur et des Vaiss. 2005, 98, 449–454.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.1K
Entry Collection:
Hypertension and Cardiovascular Diseases
Revisions:
3 times
(View History)
Update Date:
10 Jun 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No