Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Massimo Donadelli | + 3382 word(s) | 3382 | 2021-06-01 08:47:24 | | | |
| 2 | Peter Tang | Meta information modification | 3382 | 2021-06-08 05:31:40 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Donadelli, M. GAPDH-Mediated Autophagy. Encyclopedia. Available online: https://encyclopedia.pub/entry/10590 (accessed on 23 July 2026).
Donadelli M. GAPDH-Mediated Autophagy. Encyclopedia. Available at: https://encyclopedia.pub/entry/10590. Accessed July 23, 2026.
Donadelli, Massimo. "GAPDH-Mediated Autophagy" Encyclopedia, https://encyclopedia.pub/entry/10590 (accessed July 23, 2026).
Donadelli, M. (2021, June 07). GAPDH-Mediated Autophagy. In Encyclopedia. https://encyclopedia.pub/entry/10590
Donadelli, Massimo. "GAPDH-Mediated Autophagy." Encyclopedia. Web. 07 June, 2021.
Copy Citation
The cytosolic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) has pleiotropic functions independent of its canonical role in glycolysis. The GAPDH functional diversity is mainly due to post-translational modifications in different amino acid residues or due to protein–protein interactions altering its localization from cytosol to nucleus, mitochondria or extracellular microenvironment. Non-glycolytic functions of GAPDH include the regulation of cell death, autophagy, DNA repair and RNA export, and they are observed in physiological and pathological conditions as cancer and neurodegenerative disorders
GAPDH
autophagy
cancer
aggregates
cell death
1. Introduction
Autophagy is a physiological degradative mechanism of the cells by which autophagic vesicles deliver unfolded proteins and damaged organelles to lysosomes for their elimination. In this way, autophagy induces the degradation and recycling of cellular components to support energy metabolism in stressing conditions and cellular homeostasis [1]. Accumulating evidence also demonstrates that autophagy declines with age and that impaired autophagy favors individuals to age-related diseases, whereas interventions that stimulate autophagy can promote longevity [2]. However, the excessive stimulation of autophagy can be considered a self-eating mechanism favoring cell death and the aberrant regulation of autophagy is widely studied to improve therapeutic treatments for various diseases including cancer [3]. In different stages of tumorigenesis, autophagy may have opposite and context-dependent roles acting as tumor suppressor or cancer promoter [4]. Many external stimuli may affect autophagy in cancer, such as hypoxia, acidification of tumor microenvironment, nutrient deprivation, drug therapies, or infections [5][6]. A global analysis of the mutational status of the genes encoding the entire core autophagy machinery reveals that it is generally not targeted by high-frequency somatic single-nucleotide mutations across cancers [7]. These large-scale analyses indicate that the core autophagy machinery largely escapes genomic mutations and cancer cells generally express a functionally intact autophagy pathway. An exception to this general scheme is represented by the tumor suppressor BECN1 (ATG6), which lost with high frequency one allele in various cancer types [8]. However, BECN1 is adjacent to the known tumor suppressor gene breast cancer 1 (BRCA1) on chromosome 17. Hereditary breast cancer commonly results from the presence of a pathogenic germline missense mutation in BRCA1 followed by somatic deletion of the remaining wild-type BRCA1 allele. These deletions are typically large, deleting BRCA1 along with hundreds of other genes, including BECN1. Further studies demonstrated that BRCA1 loss is the driver mutation in hereditary and sporadic breast cancer [9], casting doubt on BECN1′s role as a tumor suppressor. In cancers, the autophagic flux can be modulated by tumor suppressors or oncogenes. A crucial example is that of p53, the status of which can modify the role of autophagy in tumor progression [10][11]. Furthermore, several studies indicate that p53 triggers autophagy in cancer cells through various mechanisms, such as the stimulation of AMPK, the inhibition of mTOR, or the induction of the autophagy-related gene DRAM1, to react to genotoxic or environmental stimuli [12][13]. On the other side, mutant p53 proteins counteract the autophagic machinery by various molecular mechanisms, including the transcriptional repression of ATG12 [14]. Notably, mutant p53 protein stability is affected by lysosome-mediated degradation through autophagy, supporting the concept of a functional crosstalk between mutant p53 proteins and autophagy in cancer progression [15].
Curiously, even a glycolytic enzyme, i.e., glyceraldehyde-3-phosphate dehydrogenase (GAPDH), may regulate autophagy and cell death through mechanisms that we summarize in this review. In addition to the main glycolytic role of the tetrameric conformation of GAPDH, which catalyzes the reversible conversion of glyceraldehyde-3-phosphate to 1,3-bisphosphoglycerate, GAPDH serves as a versatile enzyme that plays several regulatory roles determining the fate of the cells [16]. Indeed, GAPDH performs pleiotropic functions that mainly depend on its subcellular localization [17]. In addition to the cytosolic distribution of the glycolytic enzyme, GAPDH can also be detected close to the plasma membrane, mitochondria, nucleus, endoplasmic reticulum (ER), polysomes, Golgi and even secreted in the extracellular space carrying out several key functions including the regulation of mRNA stability [18], iron uptake and transport [19], DNA repair [20], nuclear tRNA export [21], cell death [22] and intracellular membrane trafficking [23]. The GAPDH multifunctional properties can be regulated by several mechanisms, such as the formation of macromolecular complexes by protein–protein interactions; post-translational modifications, e.g., acetylation, phosphorylation and nitrosylation; or protein oligomerization. The oligomeric state of GAPDH and its propensity to aggregate is mainly dependent on various signal molecules [24]. The redox sensitive cysteine residues of the enzyme, which includes Cys-152 in the active site, are also target of reactive oxygen species (ROS) or reactive nitrogen species (RNS) and, consequently, GAPDH aggregation is influenced by several other stimuli inducing cellular oxidative/nitrosative stresses [24][25]. Besides cancer, the functional versatility of this enzyme determines that GAPDH alteration is involved in several other diseases [26] especially neurodegenerative disorders such as Alzheimer’s disease (AD), Parkinson’s disease (PD) and Huntington’s disease (HD) [27].
2. Non-Glycolytic Roles of GAPDH
Due to the non-glycolytic roles of GAPDH, this enzyme is considered a moonlight protein in the cells. These non-glycolytic roles include physio-pathological functions such as regulation of gene expression, DNA repair and replication, neurodegeneration, pathogenesis, virulence in bacteria, tubular bundling, protein–protein interactions, RNA export, as well as apoptosis and autophagy. For instance, it has been discovered that GAPDH acts as a key component of the co-activator complex of Oct-1 in the transcriptional induction of histone H2B gene during the S phase of the cell cycle. Intriguingly, GAPDH interacts directly with Oct-1 and it has an intrinsic activation domain that can relate with the general transcription machinery [28]. This proof-of-concept opened the way to the investigation of target genes which can be transcriptionally regulated by nuclear GAPDH.
Concerning autophagy, it has been demonstrated that GAPDH may also act as a glucose sensor in the cells stimulating autophagic degradation. Indeed, during glucose starvation, the AMPK-dependent GAPDH phosphorylation is essential for SIRT1 activation and stimulation of autophagy. In these conditions, cytoplasmic GAPDH is phosphorylated by activated AMPK prompting GAPDH to redistribute into the nucleus. Inside the nucleus, GAPDH directly interacts with SIRT1, displacing SIRT1’s repressor and increasing SIRT1 deacetylase activity [29]. In general, the multiple activities of GAPDH are related to its translocation to the nucleus or to different subcellular compartments in addition to the cytosolic localization, where its main role in glycolysis is well characterized.
3. Subcellular Localization of GAPDH
The subcellular and glycolytic-independent redistribution of the enzyme can mainly occur in the nucleus, mitochondria, and intra- and extra-cellular vesicles. The regulation of GAPDH compartmentalization is mainly due to its oligomerization, post-transcriptional modifications and protein–protein interactions, thus defining also the non-glycolytic functions of GAPDH. These findings are schematically represented in Figure 1.
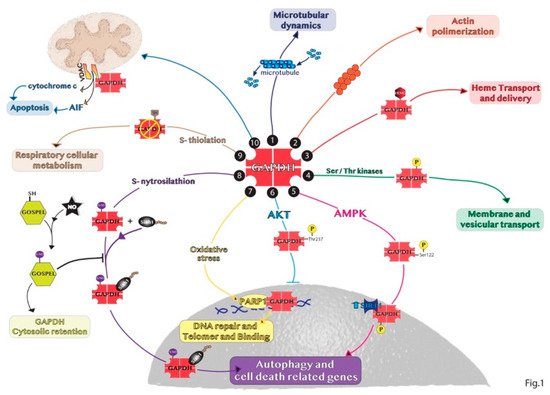
Figure 1. Representative summary of the non-glycolytic roles of GAPDH. (1) GAPDH interaction with microtubule helps in microtubular dynamics. (2) GAPDH favors actin polymerization. (3) GAPDH acts as chaperone with endogenous and exogenous heme. (4) GAPDH phosphorylation is involved in early secretory pathway transport. (5) In response to glucose deprivation, activated AMPK phosphorylates GAPDH in the cytosol inducing GAPDH translocation to the nucleus and its binding with SIRT1 promoting autophagy. (6) GAPDH phosphorylation by AKT blocks the translocation of the enzyme to the nucleus. (7) In the nucleus, GAPDH binds to PARP1 or damaged DNA to induce DNA repair. (8) SNO-GAPDH binds Siah1 and the complex translocates into the nucleus inducing cell death related genes. (9) GAPDH glycolytic activity can be inhibited by S-thiolation increasing respiratory cellular metabolism. (10) GAPDH binds VDAC1 channel favoring apoptosis by releasing cytochrome c and AIF.
4. The Double Role of Autophagy in Cancer
Autophagy is a highly conserved catabolic mechanism that involves the formation of double-membrane vesicles, the autophagosomes, by which cellular materials are delivered to lysosomes for degradation and recycled into metabolic and biosynthetic pathways. Cytoplasmic materials are delivered to the lysosome through various type of autophagy: macroautophagy, microautophagy and chaperone-mediated autophagy (CMA). Among these various types of autophagy, macroautophagy (autophagy hereafter) is the most extensively analyzed and the major catabolic mechanism used by eukaryotic cells to maintain nutrient homeostasis and organellar quality control. On the contrary, CMA is not mediated by the autophagosome and the cytoplasmic substrates are recognized by chaperone proteins, such as Hsc70, and directed toward translocation into the lysosome for degradation [30].
Autophagy is mediated by a set of conserved genes. From yeast genetic studies to those on mammalian, the breakthrough in elucidation of the molecular machinery in autophagy came from the discovery of 35 autophagy-related (ATGs) genes. All ATG genes are required for the different steps of the autophagy: among them, ATG 1–10, 12–14, 16–18, 29 and 31 are essential for the efficient formation of autophagosome. The lysosomal degradation pathway is usually described as involving a set of about 16–20 core conserved genes. The ATG proteins encoded by these genes are traditionally classified into distinct biochemical and functional groups that act at specific stages of the autophagic flux [30][31]. The formation and turnover of the autophagosome is divided into five distinct stages: (i) initiation due to starvation conditions or other stress factors, during which a decrease of glucose transport results in the release of mTOR inhibition of the ULK1 complex; (ii) nucleation of the autophagosome by ULK1 and class III PI3K complexes; (iii) expansion and elongation of the autophagosome membrane mediated by two ubiquitin-like conjugation systems, the first system being represented by LC3I/PE and LC3II complex, while the second one involving ATG5-ATG12 conjugate mediated by ATG7 and ATG10 genes; (iv) closure and autophagosome fusion with lysosome to form an autophagolysosome by the SNARE protein syntaxin 17 (STX17); and (v) degradation of intravesicular products due to the low pH of the lysosome [32].
Autophagy has opposite and context-dependent roles in cancer: under certain circumstances, autophagy may be detrimental either via its prosurvival effects or via possible cell-death promoting effects. Thus, the role of autophagy in cancer is complex and controversial. Autophagy was originally thought to represent only a tumor suppression mechanism since Aita et al. and Liang et al. found an allelic loss of an autophagic gene, BECN1 (ATG6), whose position was in close proximity to the tumor suppressor breast cancer 1 gene (BRCA1) [8][33].
In the early stages of neoplastic transformation, autophagy can act as a mechanism to counteract tumorigenesis by preventing the accumulation of damaged proteins and organelles and the excessive production of ROS that can promote DNA mutations and thus the development of neoplastic cells. In this way, autophagy limits oncogenic signaling and suppresses the onset of cancer. This may suggest a role for the stimulation of the autophagic process in the prevention of cancer occurrence [34]. Over the past 10 years, significant progress has been made in understanding the molecular mechanisms of autophagy and one conceptual advance is that autophagy can act also as a tumor promotion mechanism. The ability of autophagy to support cell survival under unfavorable environmental conditions, such as lack of nutrients or oxygen, which are extremely frequent in a growing tumor could help the survival of cancer cells. The tumors therefore exploit autophagy to their own advantage to promote their survival through the self-production of metabolic substrates necessary for the sustenance and spread of the tumor. Although it has been recognized that autophagy has an impact on the regulation of cell growth, the precise role of autophagy is highly contextual. Indeed, the aberrant stimulation of autophagy can determine an excessive auto-degradative event and self-eating mechanism supporting cell death.
Knocking down the expression of essential autophagy genes or deleting them can reduce tumorigenesis, confirming the functional importance of autophagy in tumor promotion. Autophagy is also upregulated in hypoxic tumor regions where it is required for tumor cell survival [35]. Thus, both the activation of cancer pathways within tumor cells and stress in the tumor microenvironment can increase the requirement for autophagy to promote tumor growth and survival [34].
Our group demonstrated that the inhibition of expression of autophagic genes by mutant p53 increases the proliferation of pancreatic cancer cells [14]. These results support the hypothesis of a new mechanism by which oncogenic mutant p53 protein promotes tumor proliferation with the concomitant inhibition of autophagy. The discovery of this double role of autophagy in human cancers has already led to the development of promising new cancer drugs so far. In another recent study, Ranieri et al. identified a new biological mechanism, which acts on the blocking of autophagic protective process and consequently inducing cell death and the reduction and elimination of the tumor [36]. In this study, they also identified a new pharmacologically active molecule, which can modulate this process resulting in a specific antitumor activity in melanoma cells.
5. GAPDH-Mediated Autophagy
The moonlight GAPDH protein is one of the regulators of autophagy. Besides playing the glycolytic role in the cytosol, GAPDH participates in several non-glycolytic functions including autophagy [37]. Various conditions exhibit a correlation between autophagy and the translocation of GAPDH in different subcellular compartments, especially to nucleus. For instance, GAPDH negatively regulates autophagy through the interaction with the key autophagy component ATG3 in plants [38][39]. Indeed, ROS affects the interaction between cytoplasmic GAPDH and ATG3, making free ATG3 proteins available for use in autophagy. In cardiomyocytes, oxidative stress conditions induce mitochondrial association with GAPDH promoting mitophagy, the selective degradation of mitochondria by autophagy. The formation of multiorganellar lysosomal-like (LL) structures for elimination of damaged mitochondria, also by GAPDH, helps cardiomyocytes to survive during reperfusion or reoxygenation-induced injury [40]. In brain, autophagic cytotoxic effect of cocaine is mediated by the nitric oxide-GAPDH signaling pathway [41]. Several molecular mechanisms may trigger or prevent GAPDH nuclear translocation to regulate autophagic events particularly in cancer. For instance, we discovered that the oncogenic mutant p53 protein prevents GAPDH-mediated autophagy in pancreatic cancer cells blocking the nuclear translocation of GAPDH through the stimulation of AKT and inhibition of AMPK signaling pathways [42][43], which are reported to directly phosphorylate GAPDH resulting in the inhibition or in the stimulation of GAPDH nuclear translocation, respectively [44][45]. Furthermore, in the same cancer type the production of ROS by inhibition of the antioxidant mitochondrial uncoupling protein 2 (UCP2) stimulates the nuclear translocation of GAPDH which promotes autophagy [46].
The molecular mechanisms involved in GAPDH-mediated autophagy have not yet been clarified. However, GAPDH is supposed to regulate autophagy through the direct interaction of regulatory proteins in the nucleus. In chaperone-mediated autophagy (CMA), oxidized GAPDH may be a specific substrate of this proteolytic pathway. This occurs in various steps, which include binding of the enzyme to the lysosomal membrane through the interaction with monomeric LAMP-2A (lysosome-associated membrane protein type 2A), uptake into lysosomal matrix, and degradation inside this cellular compartment [47][48]. Both heat shock cognate proteins, hsc70 and hsp90, play critical roles in the dynamics of these steps.
In low-glucose conditions, GAPDH stimulates autophagy by different pathways. One is the inhibition of mTOR signaling by the interaction between GAPDH and the Ras superfamily of GTPases Rheb, preventing Rheb binding to mTOR [49] and regulating the cross talk between glycolysis pathway and the mTORC1 pathway. In this way, GAPDH may stimulate autophagy, as mTOR inhibition causes autophagic induction [50]. In addition, under glucose deficiency GAPDH is phosphorylated by AMPK and it translocates into the nucleus, binds and activates SIRT1 deacetylase, disassociating its inhibitor DBC1 [29]. Consequently, activated SIRT1 is known to stimulate autophagy, i.e., through the deacetylation of the key autophagy component LC3 in the nucleus, which is essential for its redistribution to the cytoplasm and association with autophagic membranes [51][52]. Thus, GAPDH-mediated SIRT1 activation favors the initiation of autophagy. Interestingly, since autophagy might have a role in the promotion of cancer cell survival [4], GAPDH may act as a prosurvival factor in cancer through the induction of autophagy to support the energy consumption by rapid cell proliferation. Colell et al. showed that nuclear GAPDH protects cells from caspase-independent cell death (CICD), inducing autophagy [53]. Specifically, the nuclear function of GAPDH in protecting cells from CICD is mediated by the transcriptional up-regulation of Atg12. Since nuclear GAPDH has been involved in transcriptional regulation [28], the authors suggested that GAPDH may transcriptionally regulate ATG12 directly or indirectly. Thus, GAPDH coordinates two metabolic pathways producing ATP by glycolysis and removing damaged mitochondria by autophagy to shift the cells away from CICD [53][54]. Furthermore, Bertin et al. showed that in colon carcinoma cells the increase of GAPDH expression is sufficient to induce autophagy in vitro and in vivo [55]. Even in esophageal cancer tissues, there is a strong correlation between overexpression of GAPDH and upregulation of autophagy-related genes, like ATG12 and PIK3C3 (phosphatidylinositol 3-kinase catalytic subunit type 3) [56]. Thus, the knock-down of GAPDH was found to be sufficient to reduce autophagy and ATP levels in tumor cells [57]. Indeed, the usage of koningic acid (KA), a specific GAPDH inhibitor, inhibited autophagic flux in neuroblastoma cells [58].
In summary, the manipulation of autophagy through the regulation of the nuclear translocation of GAPDH may be an important therapeutic application in oncology. For instance, we discovered that the combined treatment with target drugs, i.e., genipin and everolimus, which are inhibitors of UCP2 and mTOR, respectively, promotes GAPDH nuclear translocation stimulating the formation of autophagic vesicles and pancreas cancer cell death [46]. On the contrary, when tumors promote autophagy as a survival mechanism, the modulation of nuclear GAPDH may be a useful target to counteract autophagy. Indeed, Guan et al. showed that GAPDH-siRNA encapsulated in nano-targeted liposomes reduces the autophagic flux in cancer cells and intriguingly favors the outcome of cancer cell drug resistance [57]. Thus, GAPDH may constitute a valuable target to modulate autophagy in cancer therapy and the main mechanisms at the basis of GAPDH-mediated autophagy regulation are schematically shown in Figure 2.
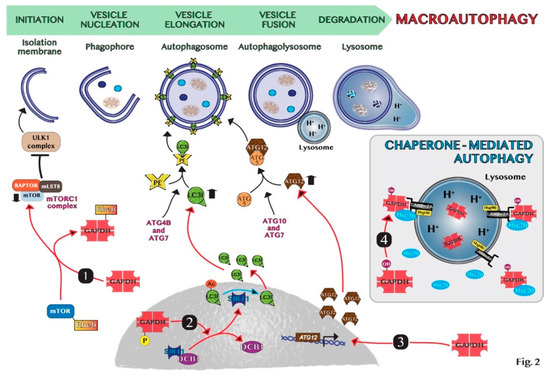
Figure 2. Schematic representation of molecular mechanisms of GAPDH-mediated autophagy: (1) GAPDH binds to Rheb, preventing Rheb binding to mTOR. (2) Phosphorylated GAPDH by AMPK translocates into the nucleus and disruptions the link between SIRT1 and DBC1. SIRT1 deacetylates LC3, which sustains autophagosome formation. (3) GAPDH enters the nucleus, favoring the induction of ATG12 gene. (4) In chaperone-mediated autophagy (CMA), GAPDH binds to LAMP-2A receptor on the lysosomal membrane. The chaperon proteins hsc70 and hsp90 are essential for CMA activity. Cytosolic Hsc70 after binding to CMA-substrates, such as GAPDH, transfers the complex to LAMP-2A. Hsp90 stabilizes LAMP-2A at the luminal side of the lysosomal membrane for the translocation of substrates.
6. Aggregation Mechanisms of GAPDH and Impact on Diseases
Protein aggregation is another process of the multiple functions involving GAPDH [59]. The high-resolution structure of the enzyme shows a highly stable tetrameric protein with identical subunits, each constituting an active site [60]. Tetrameric dissociation of the enzyme produces dimers and monomers that aggregate or bind to other biomolecules such as proteins and nucleic acids [16][61]. The apoform of the enzyme without its cofactors NAD+ or NADH, is susceptible to denaturation and consequently aggregation. These events are dependent on the content of cofactors and other ligands and on the presence of different signal molecules such as ROS that may influence the amyloidogenic processes through intermolecular disulfide bonds [25][59]. Indeed, GAPDH is an intracellular sensor of oxidative stress as discussed above. Some cysteine residues of the active site of the enzyme are strongly susceptible to various types of oxidation, decreasing the enzyme affinity to the cofactor and promoting its dissociation. The active site Cys152 of GAPDH is crucial for its aggregation induced by nitric oxide [62]. Furthermore, the site-directed mutagenesis experiments revealed that Cys149 is one of the most aggregate-prone cysteine residue of GAPDH induced by oxidative stress [25]. Several pieces of evidence show the involvement of GAPDH in the development of neurodegenerative disorders such as Alzheimer’s, Huntington’s, and Parkinson’s diseases, which are characterized by the accumulation of protein aggregates [63][64][65]. Indeed, denatured GAPDH forms may bind soluble Aβ species yielding insoluble aggregates [66][67]. The deposition of β-amyloid proteins is one of the most distinct features in Alzheimer’s disease and results from proteolytic processing of β-amyloid precursor protein (β-APP) [67]. GAPDH binds β-APP altering the normal processing of β-APP to produce β-amyloid protein and this interaction was found in amyloid plaques from the brains of patients with AD [68][69][70]. Furthermore, GAPDH aggregates promote the formation of Lewy bodies in the brains of individuals with PD [64]. GAPDH–protein interactions also occur with spinocerebellar ataxia type-1 and spinobulbar muscular atrophy gene products [71][72][73]. In conclusion, the comprehension of the mechanisms involved in the formation of GAPDH aggregates, represented in Figure 3, may help in the understanding of the biological alterations observed in neurodegenerative diseases.
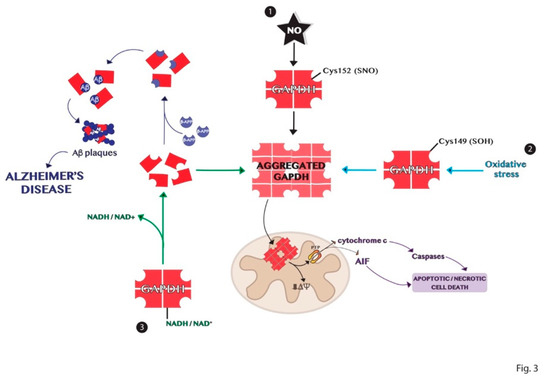
Figure 3. Schematic representation of the mechanisms of GAPDH aggregation. (1) Nitrosylation of Cys152 in the active site of GAPDH is crucial for its aggregation induced by nitric oxide. (2) Oxidation of the redox-sensitive Cys149 of GAPDH induced by oxidative stress is involved in its aggregation. (3) GAPDH without its cofactor NADH or NAD+ is susceptible to denaturation and consequent aggregation. Dimer or monomers of GAPDH can aggregate or bind other biomolecules in neurodegenerative diseases, as Alzheimer’s disease.
References
- Gao, W.; Kang, J.H.; Liao, Y.; Li, M.; Yin, X.M. Autophagy and cell death. In Essentials of Apoptosis: A Guide for Basic and Clinical Research; Yin, X.M., Dong, Z., Eds.; Humana Press: New York, NY, USA, 2009.
- Leidal, A.M.; Levine, B.; Debnath, J. Autophagy and the cell biology of age-related disease. Nat. Cell Boil. 2018, 20, 1338–1348.
- Puri, P.; Chandra, A. Autophagy Modulation As a Potential Therapeutic Target for Liver Diseases. J. Clin. Exp. Hepatol. 2014, 4, 51–59.
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967.
- Ma, Y.; Yang, H.; Pitt, J.M.; Kroemer, G.; Zitvogel, L. Therapy-induced microenvironmental changes in cancer. J. Mol. Med. 2016, 94, 497–508.
- Wojtkowiak, J.W.; Rothberg, J.M.; Kumar, V.; Schramm, K.J.; Haller, E.; Proemsey, J.B.; Lloyd, M.C.; Sloane, B.F.; Gillies, R.J.; Kumar, D.V. Chronic Autophagy Is a Cellular Adaptation to Tumor Acidic pH Microenvironments. Cancer Res 2012, 72, 3938–3947.
- Lebovitz, C.B.; Robertson, A.G.; Goya, R.; Jones, S.J.; Morin, R.D.; Marra, M.A.; Gorski, S.M. Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy 2015, 11, 1668–1687.
- Aita, V.M.; Liang, X.H.; Murty, V.; Pincus, D.L.; Yu, W.; Cayanis, E.; Kalachikov, S.; Gilliam, T.; Levine, B. Cloning and Genomic Organization of Beclin 1, a Candidate Tumor Suppressor Gene on Chromosome 17q21. Genomics 1999, 59, 59–65.
- Laddha, S.V.; Ganesan, S.; Chan, C.S.; White, E. Mutational Landscape of the Essential Autophagy Gene BECN1 in Human Cancers. Mol. Cancer Res. 2014, 12, 485–490.
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; Mackay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nat. Cell Boil. 2013, 504, 296–300.
- Yang, A.; RajeshKumar, N.V.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; Von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014, 4, 905–913.
- Comel, A.; Sorrentino, G.; Capaci, V.; Del Sal, G. The cytoplasmic side of p53’s oncosuppressive activities. FEBS Lett. 2014, 588, 2600–2609.
- Crighton, D.; Wilkinson, S.; Ryan, K.M. DRAM Links Autophagy to p53 and Programmed Cell Death. Autophagy 2007, 3, 72–74.
- Cordani, M.; Oppici, E.; Dando, I.; Butturini, E.; Pozza, E.D.; Nadal-Serrano, M.; Oliver, J.O.; Roca, P.; Mariotto, S.; Cellini, B.; et al. Mutant p53 proteins counteract autophagic mechanism sensitizing cancer cells to mTOR inhibition. Mol. Oncol. 2016, 10, 1008–1029.
- Cordani, M.; Butera, G.; Pacchiana, R.; Donadelli, M. Molecular interplay between mutant p53 proteins and autophagy in cancer cells. Biochim. Biophys. Acta Rev. Cancer 2017, 1867, 19–28.
- Tristan, C.; Shahani, N.; Sedlak, T.W.; Sawa, A. The diverse functions of GAPDH: Views from different subcellular compartments. Cell Signal. 2011.
- Sirover, M.A. Subcellular Dynamics of Multifunctional Protein Regulation: Mechanisms of GAPDH Intracellular Translocation. J. Cell. Biochem. 2012, 113, 2193–2200.
- Ikeda, Y.; Yamaji, R.; Irie, K.; Kioka, N.; Murakami, A. Glyceraldehyde-3-phosphate dehydrogenase regulates cyclooxygenase-2 expression by targeting mRNA stability. Arch. Biochem. Biophys. 2012, 528, 141–147.
- Raje, C.I.; Kumar, S.; Harle, A.; Nanda, J.S.; Raje, M. The macrophage cell surface glyceraldehyde-3-phosphate dehydrogenase is a novel transferrin receptor. J. Biol. Chem. 2007.
- Azam, S.; Jouvet, N.; Jilani, A.; Vongsamphanh, R.; Yang, X.; Yang, S.; Ramotar, D. Human Glyceraldehyde-3-phosphate Dehydrogenase Plays a Direct Role in Reactivating Oxidized Forms of the DNA repair enzyme APE1. J. Boil. Chem. 2008, 283, 30632–30641.
- Sirover, M.A. On the functional diversity of glyceraldehyde-3-phosphate dehydrogenase: Biochemical mechanisms and regulatory control. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1810, 741–751.
- Tarze, A.; Deniaud, A.; Le Bras, M.; Maillier, E.; Molle, D.; Larochette, N.; Zamzami, N.; Jan, G.; Kroemer, G.; Brenner, C. GAPDH, a novel regulator of the pro-apoptotic mitochondrial membrane permeabilization. Oncogene 2007.
- Tisdale, E.J.; Talati, N.K.; Artalejo, C.R.; Shisheva, A. GAPDH binds Akt to facilitate cargo transport in the early secretory pathway. Exp. Cell 2016, 349, 310–319.
- Gerszon, J.; Rodacka, A. Oxidatively modified glyceraldehyde-3-phosphate dehydrogenase in neurodegenerative processes and the role of low molecular weight compounds in counteracting its aggregation and nuclear translocation. Cancer Rev. 2018, 48, 21–31.
- Nakajima, H.; Amano, W.; Fujita, A.; Fukuhara, A.; Azuma, Y.-T.; Hata, F.; Inui, T.; Takeuchi, T. The Active Site Cysteine of the Proapoptotic Protein Glyceraldehyde-3-phosphate Dehydrogenase Is Essential in Oxidative Stress-induced Aggregation and Cell Death. J. Boil. Chem. 2007, 282, 26562–26574.
- Mazzola, J.L.; Sirover, M.A. Reduction of glyceraldehyde-3-phosphate dehydrogenase activity in Alzheimer’s disease and in Huntington’s disease fibroblasts. J. Neurochem. 2001, 76, 442–449.
- Chuang, D.-M.; Hough, C.; Senatorov, V.V. Glyceraldehyde-3-phosphate dehydrogenase, apoptosis, and neurodegenerative diseases. Annu. Pharmacol. Toxicol. 2005, 45, 269–290.
- Zheng, L.; Roeder, R.G.; Luo, Y. S Phase Activation of the Histone H2B Promoter by OCA-S, a Coactivator Complex that Contains GAPDH as a Key Component. Cell 2003, 114, 255–266.
- Chang, C.; Su, H.; Zhang, D.; Wang, Y.; Shen, Q.; Liu, B.; Huang, R.; Zhou, T.; Peng, C.; Wong, C.C.; et al. AMPK-Dependent Phosphorylation of GAPDH Triggers Sirt1 Activation and Is Necessary for Autophagy upon Glucose Starvation. Mol. Cell 2015, 60, 930–940.
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The Role of Atg Proteins in Autophagosome Formation. Annu. Cell Dev. Boil. 2011, 27, 107–132.
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42.
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting Autophagy in Cancer. Nat. Rev. Cancer 2017, 17, 528–542.
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nat. Cell Boil. 1999, 402, 672–676.
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, metabolism, and cancer HHS public access. Clin. Cancer Res. 2015, 21, 5037–5046.
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64.
- Ranieri, R.; Ciaglia, E.; Amodio, G.; Picardi, P.; Proto, M.C.; Gazzerro, P.; Laezza, C.; Remondelli, P.; Bifulco, M.; Pisanti, S. N6-isopentenyladenosine dual targeting of AMPK and Rab7 prenylation inhibits melanoma growth through the impairment of autophagic flux. Cell Death Differ. 2018.
- Colell, A.; Green, D.R.; Ricci, J.-E. Novel roles for GAPDH in cell death and carcinogenesis. Cell Death Differ. 2009, 16, 1573–1581.
- Henry, E.; Fung, N.; Liu, J.; Drakakaki, G.; Coaker, G. Beyond Glycolysis: GAPDHs Are Multi-functional Enzymes Involved in Regulation of ROS, Autophagy, and Plant Immune Responses. PLOS Genet. 2015, 11, e1005199.
- Han, S.; Wang, Y.; Zheng, X.; Jia, Q.; Zhao, J.; Bai, F.; Hong, Y.; Liu, Y. Cytoplastic Glyceraldehyde-3-Phosphate Dehydrogenases Interact with ATG3 to Negatively Regulate Autophagy and Immunity in Nicotiana benthamiana. Cancer Cell 2015, 27, 1316–1331.
- Yogalingam, G.; Hwang, S.; Ferreira, J.C.B.; Mochly-Rosen, D. Glyceraldehyde-3-phosphate Dehydrogenase (GAPDH) Phosphorylation by Protein Kinase Cδ (PKCδ) Inhibits Mitochondria Elimination by Lysosomal-like Structures following Ischemia and Reoxygenation-induced Injury*. J. Boil. Chem. 2013, 288, 18947–18960.
- Guha, P.; Harraz, M.M.; Snyder, S.H. Cocaine elicits autophagic cytotoxicity via a nitric oxide-GAPDH signaling cascade. Proc. Natl. Acad. Sci. USA 2015, 1–6.
- Butera, G.; Pacchiana, R.; Mullappilly, N.; Margiotta, M.; Bruno, S.; Conti, P.; Riganti, C.; Donadelli, M. Mutant p53 prevents GAPDH nuclear translocation in pancreatic cancer cells favoring glycolysis and 2-deoxyglucose sensitivity. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1914–1923.
- Cordani, M.; Butera, G.; Dando, I.; Torrens-Mas, M.; Butturini, E.; Pacchiana, R.; Oppici, E.; Cavallini, C.; Gasperini, S.; Tamassia, N.; et al. Mutant p53 blocks SESN1/AMPK/PGC-1α/UCP2 axis increasing mitochondrial O2ˉ· production in cancer cells. Br. J. Cancer 2018, 119, 994–1008.
- Huang, Q.; Lan, F.; Zheng, Z.; Xie, F.; Han, J.; Dong, L.; Xie, Y.; Zheng, F. Akt2 Kinase Suppresses Glyceraldehyde-3-phosphate Dehydrogenase (GAPDH)-mediated Apoptosis in Ovarian Cancer Cells via Phosphorylating GAPDH at Threonine 237 and Decreasing Its Nuclear Translocation*. J. Boil. Chem. 2011, 286, 42211–42220.
- Kwon, H.J.; Rhim, J.H.; Jang, I.-S.; Kim, G.-E.; Park, S.C.; Yeo, E.-J. Activation of AMP-activated protein kinase stimulates the nuclear localization of glyceraldehyde 3-phosphate dehydrogenase in human diploid fibroblasts. Exp. Mol. Med. 2010, 42, 254–269.
- Dando, I.; Pacchiana, R.; Pozza, E.D.; Cataldo, I.; Bruno, S.; Conti, P.; Cordani, M.; Grimaldi, A.; Butera, G.; Caraglia, M.; et al. UCP2 inhibition induces ROS/Akt/mTOR axis: Role of GAPDH nuclear translocation in genipin/everolimus anticancer synergism. Radic. Boil. Med. 2017, 113, 176–189.
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M.; Lippincott-Schwartz, J. Activation of Chaperone-mediated Autophagy during Oxidative Stress. Mol. Boil. Cell 2004, 15, 4829–4840.
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The Chaperone-Mediated Autophagy Receptor Organizes in Dynamic Protein Complexes at the Lysosomal Membrane. Mol. Cell. Boil. 2008, 28, 5747–5763.
- Lee, M.N.; Ha, S.H.; Kim, J.; Koh, A.; Lee, C.S.; Jeon, H.; Kim, D.-H.; Suh, P.-G.; Ryu, S.H.; Kim, J.H.; et al. Glycolytic Flux Signals to mTOR through Glyceraldehyde-3-Phosphate Dehydrogenase-Mediated Regulation of Rheb. Mol. Cell. Boil. 2009, 29, 3991–4001.
- Jung, C.H.; Ro, S.-H.; Cao, J.; Otto, N.M.; Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295.
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Acad. Sci. 2008, 105, 3374–3379.
- Huang, R.; Xu, Y.; Wan, W.; Shou, X.; Qian, J.; You, Z.; Liu, B.; Chang, C.; Zhou, T.; Lippincott-Schwartz, J.; et al. Deacetylation of Nuclear LC3 Drives Autophagy Initiation under Starvation. Mol. Cell 2015, 57, 456–466.
- Colell, A.; Ricci, J.-E.; Tait, S.; Milasta, S.; Maurer, U.; Bouchier-Hayes, L.; Fitzgerald, P.; Guio-Carrion, A.; Waterhouse, N.J.; Li, C.W.; et al. GAPDH and Autophagy Preserve Survival after Apoptotic Cytochrome c Release in the Absence of Caspase Activation. Cell 2007, 129, 983–997.
- Song, S.; Finkel, T. GAPDH and the search for alternative energy. Nat. Cell Boil. 2007, 9, 869–870.
- Bertin, S.; Samson, M.; Pons, C.; Gavelli, A.; Baqué, P.; Brossette, N.; Pagnotta, S.; Pierrefite-Carle, V.; Guigonis, J.-M.; Ricci, J.-E. Comparative Proteomics Study Reveals That Bacterial CpG Motifs Induce Tumor Cell Autophagy in Vitro and in Vivo*S. Mol. Cell. Proteom. 2008, 7, 2311–2322.
- Soltany-Rezaee-Rad, M.; Mottaghi-Dastjerdi, N.; Setayesh, N.; Roshandel, G.; Ebrahimifard, F.; Sepehrizadeh, Z. Overexpression of FOXO3, MYD88, and GAPDH Identified by Suppression Subtractive Hybridization in Esophageal Cancer Is Associated with Autophagy. Gastroenterol. Pr. 2014, 2014, 1–8.
- Guan, J.; Sun, J.; Sun, F.; Lou, B.; Zhang, D.; Mashayekhi, V.; Sadeghi, N.; Storm, G.; Mastrobattista, E.; He, Z. Hypoxia-induced tumor cell resistance is overcome by synergistic GAPDH-siRNA and chemotherapy co-delivered by long-circulating and cationic-interior liposomes. Nanoscale 2017, 9, 9190–9201.
- Dodson, M.; Liang, Q.; Johnson, M.S.; Redmann, M.; Fineberg, N.; Darley-Usmar, V.M.; Zhang, J. Inhibition of glycolysis attenuates 4-hydroxynonenal-dependent autophagy and exacerbates apoptosis in differentiated SH-SY5Y neuroblastoma cells. Autophagy 2013, 9, 1996–2008.
- Muronetz, V.I.; Barinova, K.V.; Stroylova, Y.Y.; Semenyuk, P.I.; Schmalhausen, E.V.; Barinova, K.V. Glyceraldehyde-3-phosphate dehydrogenase: Aggregation mechanisms and impact on amyloid neurodegenerative diseases. Int. J. Boil. Macromol. 2017, 100, 55–66.
- Sirover, M.A. Role of the glycolytic protein, glyceraldehyde-3-phosphate dehydrogenase, in normal cell function and in cell pathology. J. Cell. Biochem. 1997, 66, 133–140.
- I Arutyunova, E.; Danshina, P.V.; Domnina, L.V.; Pleten, A.P.; I Muronetz, V. Oxidation of glyceraldehyde-3-phosphate dehydrogenase enhances its binding to nucleic acids. Biochem. Biophys. Commun. 2003, 307, 547–552.
- Kubo, T.; Nakajima, H.; Nakatsuji, M.; Itakura, M.; Kaneshige, A.; Azuma, Y.-T.; Inui, T.; Takeuchi, T. Active site cysteine-null glyceraldehyde-3-phosphate dehydrogenase (GAPDH) rescues nitric oxide-induced cell death. Nitric Oxide 2016, 53, 13–21.
- Wang, Q.; Woltjer, R.L.; Cimino, P.J.; Pan, C.; Montine, K.S.; Zhang, J. Proteomic analysis of neurofibrillary tangles in Alzheimer disease identifies GAPDH as a detergent-insoluble paired helical filament tau binding protein. FASEB J. 2005, 19, 869–871.
- Tsuchiya, K.; Tajima, H.; Kuwae, T.; Takeshima, T.; Nakano, T.; Tanaka, M.; Sunaga, K.; Fukuhara, Y.; Nakashima, K.; Ohama, E.; et al. Pro-apoptotic protein glyceraldehyde-3-phosphate dehydrogenase promotes the formation of Lewy body-like inclusions. Eur. J. Neurosci. 2005, 21, 317–326.
- Bae, B.-I.; Hara, M.R.; Cascio, M.B.; Wellington, C.L.; Hayden, M.R.; Ross, C.A.; Ha, H.C.; Li, X.-J.; Snyder, S.H.; Sawa, A. Mutant Huntingtin: Nuclear translocation and cytotoxicity mediated by GAPDH. Proc. Acad. Sci. 2006, 103, 3405–3409.
- Verdier, Y.; Foldi, I.; Sergeant, N.; Fülöp, L.; Penke, Z.; Janáky, T.; Szücs, M.; Penke, B. Characterization of the interaction between Aβ 1–42 and glyceraldehyde phosphodehydrogenase. J. Pept. Sci. 2008, 14, 755–762.
- Selkoe, D.J. Alzheimer’s Disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–767.
- Butterfield, D.A.; Hardas, S.S.; Lange, M.L.B. Oxidatively modified glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Alzheimer’s disease: Many apathways to neurodegenertion. J. Alzheimer’s Dis. 2010.
- Mazzola, J.L.; Sirover, M.A. Alteration of nuclear glyceraldehyde-3-phosphate dehydrogenase structure in Huntington’s disease fibroblasts. Mol. Brain 2002, 100, 95–101.
- Cumming, R.C.; Schubert, D. Amyloid-β induces disulfide bonding and aggregation of GAPDH in Alzheimer’s disease. FASEB J. 2005, 19, 2060–2062.
- Koshy, B.; Matilla, T.; Burright, E.N.; Merry, D.E.; Fischbeck, K.H.; Orr, H.T.; Zoghbi, H.Y. Spinocerebellar ataxia type-1 and spinobulbar muscular atrophy gene products interact with glyceraldehyde-3-phosphate dehydrogenase. Hum. Mol. Genet. 1996, 5, 1311–1318.
- Burke, J.R.; Enghild, J.J.; Martin, M.E.; Jou, Y.-S.; Myers, R.M.; Roses, A.D.; Vance, J.M.; Strittmatter, W.J. Huntingtin and DRPLA proteins selectively interact with the enzyme GAPDH. Nat. Med. 1996, 2, 347–350.
- Shiozawa, M.; Fukutani, Y.; Arai, N.; Cairns, N.J.; Mizutani, T.; Isaki, K.; Lantos, P.L.; Wada, Y. Glyceraldehyde 3-phosphate dehydrogenase and endothelin-1 immunoreactivity is associated with cerebral white matter damage in dentatorubral-pallidoluysian atrophy. Neuropathology 2003, 23, 36–43.
More
Information
Subjects:
Pathology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.3K
Revisions:
2 times
(View History)
Update Date:
08 Jun 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No