Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Francesco Napolitano | + 3332 word(s) | 3332 | 2021-05-31 05:43:17 | | | |
| 2 | Peter Tang | Meta information modification | 3332 | 2021-06-08 05:30:15 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Napolitano, F. Rhes in Parkinson’s Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/10588 (accessed on 28 June 2026).
Napolitano F. Rhes in Parkinson’s Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/10588. Accessed June 28, 2026.
Napolitano, Francesco. "Rhes in Parkinson’s Disease" Encyclopedia, https://encyclopedia.pub/entry/10588 (accessed June 28, 2026).
Napolitano, F. (2021, June 07). Rhes in Parkinson’s Disease. In Encyclopedia. https://encyclopedia.pub/entry/10588
Napolitano, Francesco. "Rhes in Parkinson’s Disease." Encyclopedia. Web. 07 June, 2021.
Copy Citation
Rhes is one of the most interesting genes regulated by thyroid hormones that, through the inhibition of the striatal cAMP/PKA pathway, acts as a modulator of dopamine neurotransmission. Rhes mRNA is expressed at high levels in the dorsal striatum, with a medial-to-lateral expression gradient reflecting that of both dopamine D2 and adenosine A2A receptors. Rhes transcript is also present in the hippocampus, cerebral cortex, olfactory tubercle and bulb, substantia nigra pars compacta (SNc) and ventral tegmental area of the rodent brain. In line with Rhes-dependent regulation of dopaminergic transmission, data showed that lack of Rhes enhanced cocaine- and amphetamine-induced motor stimulation in mice.
substantia nigra
mTOR
SUMO E3 ligase
Huntington’s disease
3
4-methylenedioxymethamphetamine (MDMA)
autophagy
L-Dopa-induced dyskinesia (LID)
mitophagy
1. Discovery of Rhes
1.1. Protein Structure
The Ras homolog enriched in striatum (Rhes) is a 266 amino-acid (aa) protein, discovered by a subtractive hybridization procedure, in the attempt to identify striatal-enriched transcripts [1]. As the name implies, Rhes belongs to the superfamily of Ras proteins and, as such, it is made up of five G box domains, all of them normally required for the interaction with phosphate moieties of guanosine triphosphate/diphosphate (GTP/GDP) Ras-GTPase activating protein effector, and guanine nucleotide moiety [2]. Together with Dexras1, Rhes differs from other cognate members for having peculiar N- and C-terminal domains [3][4]. In this respect, while the N-terminal sequence, encompassing 1–18 amino acids, is likely to have the binding motif for the deubiquitinating enzyme, the C-terminal cationic domain interacts with Gβ1, Gβ2 and Gβ3 subunits of heterotrimeric G proteins [5], and contains a well-conserved CAAX motif that, following the enzymatic post-translational modification (farnesylation), is able to translocate this small protein to the plasma membrane [6][7][8].
1.2. Anatomical Brain Localization
Rhes mRNA was detected in virtually all GABAergic medium spiny projection neurons (MSNs) of rodent and human brains, as well as in mouse large aspiny cholinergic interneurons (ChIs), but not in GABAergic parvalbumin- and neuropeptide Y-positive interneurons of the mouse striatum [9][10][11]. The expression of Rhes was reported to be higher in the dorsal striatum than the ventral striatum (nucleus accumbens), with a peculiar medial-to-lateral gradient of increasing expression observed in both young (from 6-day-old) and adult rodents [10][12][13], thus mirroring the striatal expression pattern of both dopamine D2 receptor (D2R) and adenosine A2A receptor (A2AR) as well [10][14]. In addition to the initial studies about its striatal localization, Rhes mRNA was also detected in several other areas of the central nervous system, such as the cornu Ammonis (CA) of the hippocampus (i.e., CA1, CA2 and CA3 subfields), cerebral cortex (layers II and III), piriform cortex, olfactory tubercle, subiculum, thalamus, inferior colliculus, substantia nigra pars compacta (SNc) and ventral tegmental area (VTA) of the rodent brain [4][13][15]. Similarly, in the human brain, Rhes transcript was observed in the hippocampal dentate gyrus and in the pyramidal cell layer of CA1, CA2 and CA3 fields [10], as well as in frontal cortical areas (layers II–VI), with the highest expression observed in layer V of the cerebral cortex [16]. More detailed studies, somehow supporting and extending such findings, were recently performed by Ehrenberg and colleagues, who documented that, using multiplex immunofluorescence and single nucleus RNA-sequencing approaches in human brain, Rhes is widespread in cortical neurons, CA1 pyramidal neurons, superior frontal gyrus and entorhinal cortex, where it presents an almost total diffuse cytoplasmic distribution [6].
2. Ontogeny of Rhes and Its Striatal Regulation
2.1. Rhes Is Modulated by Thyroid Hormones
The first gene expression study aimed to evaluate the ontogeny of Rhes mRNA in rats was carried out by Falk and colleagues in the 1999, who documented low levels of between embryonic day 16 (E16) and postnatal day 10 (P10), while a seven-fold increase occurred between P10 and P15 [7], and stabilized from that time on [13]. This peculiar Rhes expression pattern mirrors that of thyroid hormones’ occurrence and prompted researchers to investigate about the putative functional correlation between Rhes and thyroid hormones. In this respect, Northern blot and in situ hybridization analyses, carried out in the striatal samples of congenital hypothyroid rats, revealed levels of Rhes mRNA as barely detectable, which were normalized following the physiological thyroxine (T4) supplementation, either by a single or repeated 3,3′,5-triiodo-L-thyronine (T3) injections [7][17][18][19]. Interestingly, no Rhes transcript changes were observed in adult onset of hypothyroidism in rats [19], whereas adult hypothyroid mice showed a significant reduction in striatal Rhes transcript [18]. Again, administration of the selective thyroid hormone receptor-beta (TRβ) agonist GC-1 was able to normalize striatal Rhes mRNA in congenitally hypothyroid 17-day-old rats, suggesting a significant contribution of TRβ in Rhes expression [17]. However, a later study in mice highlighted a major role for thyroid hormone receptor-alpha (TRα), as T3 supplementation was able to rescue striatal Rhes transcript exclusively in TRβ-deficient animals, but not in TRα-deficient ones [18].
2.2. Rhes Expression Is Regulated by Dopamine Innervation
Besides thyroid hormones, other evidence outlined a role played by dopamine innervation in regulating striatal Rhes mRNA in adult rodents. Accordingly, dopamine depletion, induced either by the dopaminergic/noradrenergic neurotoxin 6-hydroxydopamine (6-OHDA) or reserpine, significantly reduced Rhes mRNA levels throughout the striatum and olfactory tubercle of adult rats [12], while no main effect was observed in 6-OHDA-lesioned neonatal animals [13]. Consistent with observations drawn from rodents, Napolitano and coworkers also reported a significant reduction of Rhes mRNA levels in the striatum of both 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-intoxicated non-human primates (Macaca mulatta) and Parkinson’s disease (PD) patients [20]. Overall, these findings suggest a link between intact dopaminergic innervation and physiological Rhes mRNA expression and, in turn, unveil a potential involvement of this small GTPase in PD pathophysiology.
3. Rhes Intracellular Signaling
3.1. In Vitro Modulation of Rhes-Dependent cAMP/PKA Signaling
The first insight about biochemical properties of Rhes was provided by Vargiu and collaborators (2004), who documented that in undifferentiated PC12 cells, Rhes seems to be active even under resting conditions, although with a low intrinsic GTPase activity, since more than 30% of this protein resulted bound to GTP [4]. Moreover, the same authors found that co-transfection of Rhes, either with thyrotropin-stimulating hormone receptor (TSHR), or with constitutively activated β2-adrenergic receptors, significantly inhibited the cyclic adenosine monophosphate (cAMP)/phosphate kinase A (PKA) activity. Of interest, Rhes did not directly interfere with the function of either Gαs/olf protein or PKA, suggesting an upstream site of action, most likely between GPCR localization and heterotrimeric G protein complex. In agreement with this view, it was later reported the ability of Rhes to affect in vitro the drug-stimulated activation of the dopamine type 1 receptor (D1R), with a significant reduction of cAMP accumulation and the downstream-related signaling [21]. Alongside its ability to negatively modulate GPCR signaling, further experiments in HEK293 and COS-7 cells showed that Rhes reduces Gαi-dependent signaling, by inhibiting tonic voltage-dependent CaV2.2 (N-type) calcium channels, in a pertussis toxin (PTX)-dependent manner [21][22].
3.2. Rhes Affects Striatal cAMP/PKA Signaling in Mice
Consistent with observation performed in vitro, a negative modulatory role of Rhes over striatal D1R-dependent cAMP/PKA signaling in mice was reported. In this respect, administration of SKF 81297, a selective dopamine D1R agonist, caused a greater increased phosphorylation state of the PKA-dependent activation site Ser-845 residue of the glutamate α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor subunit, in Rhes knockout (KO) mice, when compared to wild-type (WT) controls [9][10]. Besides D1R, Rhes can also counteract striatal D2R-mediated signaling, as demonstrated by the reduced ability for dopamine to activate Gi/o protein, in striatal slices from Rhes KO mice [9]. In line with the signaling properties of Rhes in regulating dopaminergic transmission, more recent investigations showed that lack of Rhes significantly enhanced amphetamine-induced motor stimulation in KO mice, most likely also through the inhibitory control of the striatal-enriched guanine nucleotide exchange factor (GEF), RasGRP1, over Rhes activity [16][23]. In keeping with this, Napolitano and colleagues showed that Rhes profoundly impacted on molecular and motor stimulant effects mediated by cocaine administration. Indeed, mice lacking the Rhes gene showed an abnormally higher motor response to this psychostimulant in Rhes KO mice than WT-treated animals. Moreover, remarkable changes in cocaine-dependent protein expression were reported in KO animals within whole striatal proteome, when compared to controls [24]. Altogether, these results suggest that Rhes might act as a physiological molecular brake for the striatal dopamine responses, under phasic conditions [20][25].
3.3. Rhes Affects the PI3K/Akt Signaling Pathway
Early experiments, carried out in HeLa cells, indicated that Rhes functionally binds to the catalytic p110 subunit of PI3K, and when co-transfected in Cos-7 cells with Akt, it promotes Akt-mediated phosphorylation of histone H2B [4]. These findings were later confirmed and extended in HEK293T and PC12 cells where, following the treatment with different growth factors (IGF-1, EGF or PDGF), Rhes enhanced p85-PI3K interaction and, interestingly, targeted Akt to the plasma membrane, thus arguing that Rhes may function as a critical bridge between PI3K and the AKT pathway [26]. In line with in vitro data, lack of Rhes results in profound alteration in the excitability of ChIs, where the stimulation of D2R triggered an aberrant increase of action potential discharge, which was prevented by the pre-incubation with either the selective CaV2.2 Ca2+ channel blocker, ω-conotoxin, or PI3K inhibitor, LY294002, pointing towards a functional modulation of Rhes on the PI3K/Akt signaling pathway in these neurons [11]. On the other hand, in vivo studies performed in Rhes KO mice demonstrated that lack of Rhes increased phosphorylation of Akt and glycogen synthase kinase 3 beta (GSK3-β), upon apomorphine treatment, assuming that this small molecule may be necessary to promote Akt dephosphorylation [27]. Moreover, the same authors documented that Rhes interacts with β-arrestin [27], a scaffolding protein, which is established to modulate the D2R-dependent Akt/GSK3-β signaling [28].
4. Rhes Involvement in Huntington Disease and L-DOPA-Induced Dyskinesia
4.1. Rhes Acts as SUMO E3 Ligase for the Mutant Huntingtin
Small ubiquitin-like modifier proteins (SUMO) represent a category of molecules covalently attached to specific lysine target residues, thus allowing changes in their localization, stability and activity, by means of a dynamic process, known as SUMOylation [29]. Interestingly, given its relevant impact on the modulation of synaptic plasticity, SUMOylation has also been implicated in a variety of neurological disorders, including PD, Huntington’s disease (HD) and amyotrophic lateral sclerosis (for a review, refer to Anderson et al., 2017 [30]). In this respect, compelling evidence pointed out that Rhes acts as SUMO E3 ligase in the striatum and, by doing so, it may participate in the HD pathogenesis, as well as in tau pathology [8][31][32]. Specifically, it was demonstrated the ability of Rhes to less avidly bind to WT huntingtin (wtHtt), and drastically increase the disperse (cytotoxic) form of mutant huntingtin (mHtt), as compared to the aggregated (cytoprotective) one, in different cellular settings [32][33]. Additionally, Rhes participates in the SUMOylation process throughout the striatum, by promoting the “cross-SUMOylation” of E1 and Ubc9 (E2) proteins, thus influencing several signaling pathways [34].
4.2. Role of Rhes in Modulating HD-Dependent Phenotypes in Animal Models
In agreement with the above-mentioned in vitro findings, in vivo evidence strengthened the potential involvement of Rhes in HD, since lack of Rhes prevented the striatal injury and motor dysfunctions in Rhes KO mice, induced by the mitochondrial complex II inhibitor, 3-nitropropionic acid (3-NP) [35]. Moreover, Rhes gene deletion either delayed or ameliorated behavioral and anatomical HD-related phenotypes in the transgenic mouse models of HD, R6/1 and B6.129P2-Htttm2Detl/150J, which display about 115 CAG repeats of the human mHtt allele and just the N-terminal fragment of mHtt, respectively [36][37]. Interestingly, investigations performed in R6/2 and 140 CAG knock-in HD mouse models revealed that the Golgi protein acyl-CoA binding domain containing 3 (ACBD3) and the huntingtin-associated protein 1 (Hap1) oppositely modulated Rhes E3 ligase activity, either increasing or reducing Rhes-mediated SUMOylation of mHtt [38][39]. Rhes has been recently regarded as an inducer of tunneling nanotubes (TNT)-like protrusions, which allow the communication of neighboring cells, as well as transport of the selective membrane vesicles and organelles, including mHtt rather than wtHtt [40]. Accordingly, studies of differential interference contrast microscopy, carried out in the striatal STHdhQ7/Q7 cells, demonstrated that, out of 70% of GFP-Rhes-positive cells showing filopodia-like protrusions, 30% of them exhibited TNT-like structures, thus highlighting a novel ability for Rhes to modulate striatal HD vulnerability [40]. It is worth underlying that the SUMO E3 ligase activity domain of Rhes (171–266 aa) promotes the biogenesis of TNT-like tunnels, even if only the full-length Rhes WT protein can be transported from cell to cell [40].
4.3. Rhes Affects L-DOPA-Induced Dyskinesia (LID) in PD Mouse Model
Furthermore, both in vitro and in vivo experiments showed that Rhes physiologically binds to and activates the mTOR complex 1 and 2 (mTORC1 and mTORC2, respectively), in a GTP-dependent manner [23][31]. Among a variety of cellular and molecular processes, mTORC1 has been regarded as one of the master regulators of L-3,4-dihydroxyphenylalanine (L-DOPA)-induced dyskinesia (LID) in PD rodent models [41][42]. The most efficacious symptomatic treatment in PD is the dopamine replacement with the dopamine precursor, L-DOPA. However, long-term L-DOPA therapy is associated with the development of motor complications, such as LID, which severely compromise the beneficial effects of the drug, thus becoming treatment-limiting [43][44][45][46][47]. Among different molecular changes underlying LID onset and severity [44][48], mTORC1 activation within D1-expressing striatal neurons following chronic L-DOPA treatment has been regarded as a key player in the modulation of such motor disturbances [42]. Accordingly, mTORC1 inhibition, either by rapamycin or rapamycin ester CCI-779, significantly reduced LID in 6-OHDA-lesioned PD rodent models, without affecting the anti-akinetic effect of L-DOPA [41][42]. In this view, studies in striatal cell lines, striatal tissue and HEK293 cells as well, documented that Rhes has the ability to selectively bind to and activate mTOR [49]. Remarkably, lack of Rhes significantly reduced LID occurrence and severity in 6-OHDA-lesioned KO mice, and prevented the rise of nigral GABA and glutamate release in the substantia nigra pars reticulata (SNr), which represents the output nucleus of the basal ganglia [49][50]. More recently, a direct influence of Rhes on RasGRP1-dependent signaling in promoting LID expression has been reported in animal models [51]. Overall, considering, on one hand, the potential toxicity of rapamycin and related drugs as inhibitors of protein synthesis and, on the other, the negligible levels of Rhes in peripheral tissues, these findings pave the way toward a potential use of Rhes inhibitors in PD therapy to counteract LID, with no detrimental impact on L-DOPA efficacy (Figure 1). However, in order to potentially translate this therapeutic strategy to PD patients, further new studies in Rhes conditional knockout mice, aimed at selectively deleting the Rhes gene in the nigrostriatal pathway, are mandatory, so as to better disclose the role of this small GTP-binding protein in such severe motor disturbances.
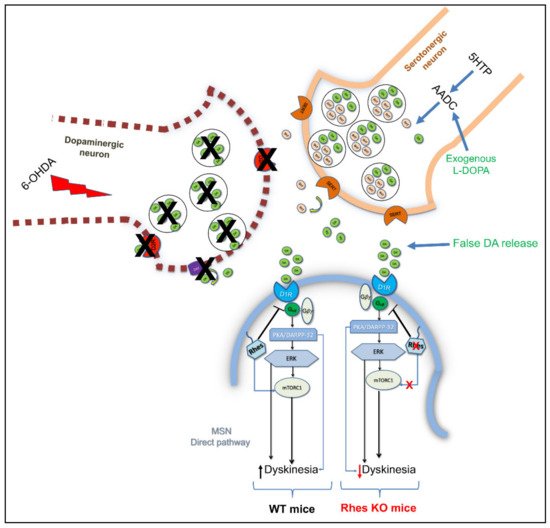
Figure 1. Rhes modulates L-DOPA-induced dyskinesia. Schematic representation showing that Rhes, following the activation of striatal mTORC1, mediates the dyskinetic effects triggered by L-DOPA administration in a 6-OHDA-lesioned mouse model, once converted to dopamine and released by serotonergic neurons in a non-physiological manner.
5. Involvement of Rhes in Parkinson’s Disease: Focus on Rhes Regulation of Nigrostriatal Neurons’ Survival
5.1. Rhes Counteracts Nigrostriatal Degeneration during Ageing in a Gender-Dependent Manner
The pathophysiology of PD relies on the degeneration of dopaminergic neurons located in the SNc (which project to the motor part of the striatum, caudate-putamen nucleus in humans), as well as cytoplasmic accumulation of α-synuclein-containing Lewy bodies [43][52]. Based on the occurrence of Rhes transcript in the midbrain tyrosine hydroxylase (TH)-positive neurons of SNc and VTA (Figure 2) [15] and, considering its role in regulating survival-related AKT and mTOR signaling pathways, further studies sought to investigate whether Rhes could also have an impact on midbrain dopaminergic neurons’ survival, under both physiological and pathological conditions. Interestingly, lack of Rhes led to a mild, although significant, reduction of midbrain TH-positive neurons in both 6- and 12-month-old KO male mice [15]. As a behavioral correlate to what was observed at the morphological level, mutant male animals showed significant alterations at the beam-walking test, in an age-dependent manner, taking longer to traverse the beam, thus suggesting that Rhes might drive the nigrostriatal pathway toward a susceptibility to cell death, triggered either by aging processes or by environmental toxins [15]. The mechanisms responsible for the PD-related neuronal degeneration are still elusive, and often controversial. However, several factors, such as gender, neuroinflammation, oxidative stress, excitotoxicity, reduced expression of trophic factors, and dysfunction of the protein degradation system, may influence the nigrostriatal pathway degeneration [53][54]. Although several causative genes of either dominant or recessive inherited PD forms have been identified, most of them are not yet characterized, hence requiring further studies aimed at clarifying the interplay between genetics and other possible pathogenic factors [55]. Yet, epidemiological investigations indicated that among the factors that may influence the neuronal dopaminergic degeneration, gender may have a prominent role, since males are at higher risk than females, who might be more protected by estrogens, in particular 17β-estradiol [56][57]. Therefore, based on the involvement of the α-synuclein-mediated microglia activation in PD pathogenesis [58][59][60], and considering the influence of Rhes upon the survival of nigrostriatal dopaminergic neurons [15], in a recent study by Costa and colleagues, the potential role of this protein on the inflammatory response was initially investigated during the physiological brain aging, in both male and female Rhes KO mice [61]. Immunohistochemistry evaluations confirm the decrease in TH immunoreactivity already observed in the midbrain nigrostriatal neurons of male Rhes KO mice [15], but a decrease in TH immunoreactivity was also observed in female Rhes KO mice. Interestingly, a higher number of the complement type 3 receptor (CD11b), as well as glial fibrillary acid protein (GFAP), were found in male rather than female KO mice [61].
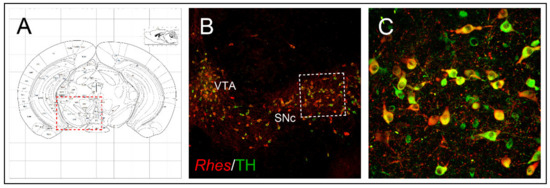
Figure 2. Rhes expression in midbrain dopaminergic neurons. (A) Schematic representation of a coronal section at the level of the midbrain. (B,C) Confocal images of brain coronal sections showing expression of Rhes in SNc and VTA TH-positive DA neurons. The figure has been adapted from Pinna et al. [15].
5.2. Rhes Reduces the MDMA-Induced Dopaminergic Degeneration and Neuroinflammation Affecting the Nigrostriatal System
Among the amphetamine-related drugs, 3,4-methylenedioxymethamphetamine (MDMA, also known as ‘ecstasy’) is one of the most heavily abused psychostimulants among adolescents and young adults [62][63][64]. MDMA has addictive properties and may elicit neurotoxic effects and glia activation in several animal species, although the impact on the neural system may differ depending on the considered species [65][66][67][68]. In particular, administration of MDMA to mice triggers a peculiar profile of neurotoxicity and glia activation that involves the nigrostriatal and mesolimbic dopamine systems [63][69][70][71][72]. These interesting results allowed us to evaluate whether Rhes KO mice showed higher vulnerability to MDMA-dependent neurotoxic and neuroinflammatory effects in the nigrostriatal system as compared to WT animals, and whether gender and/or age might be associated with these effects. In line with this, one of the studies recently performed by Usiello and co-workers demonstrated that acute-repeated MDMA administration in adult (3-month-old) and middle-aged (12-month-old) male and female Rhes KO mice caused a significant dopaminergic neurodegeneration and glia activation, which was generally more pronounced in males than females [73]. In adult males, MDMA administration induced in both WT and KO animals a decrease of TH-positive fiber density in the dorsal striatum, as well as of the total number of TH-positive neurons in SNc. Conversely, female Rhes WT and KO mice were affected by MDMA administration only with aging. In middle-aged mice, MDMA administration induced a significant decrease in the density of TH-positive fibers in the dorsal striatum and SNc in both male and female WT and Rhes KO mice. Interestingly, the decrease observed in the dorsal striatum of adult and middle-aged male Rhes KO mice was higher than that observed in WT mice. Furthermore, Rhes KO adult males showed a more pronounced astrogliosis in the dorsal striatum and microgliosis in the dorsal striatum and SNc as compared with WT and female Rhes KO animals. Finally, while adult female Rhes KO mice did not show glial activation as compared to WT, susceptibility for dopamine neuron increased with ageing, suggesting for females a lower vulnerability to neurotoxicity as compared to males. These data give support to the influence of Rhes in regulating the survival of dopaminergic neurons, as shown by Pinna et al. [15]. Overall, Rhes is able to influence the survival of the nigrostriatal pathway, making Rhes KO mice a suitable model to unveil molecular mechanisms potentially involved in the vulnerability to the midbrain dopaminergic neuronal loss, under both physiological and pathological processes.
References
- Usui, H.; Falk, J.D.; Dopazo, A.; De Lecea, L.; Erlander, M.G.; Sutcliffe, J.G. Isolation of clones of rat striatum-specific mRNAs by directional tag PCR subtraction. J. Neurosci. 1994, 14, 4915–4926.
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: Conserved structure and molecular mechanism. Nature 1991, 349, 117–127.
- Thapliyal, A.; Verma, R.; Kumar, N. Small G proteins Dexras1 and RHES and their role in pathophysiological processes. Int. J. Cell Biol. 2014, 2014, 1–10.
- Vargiu, P.; De Abajo, R.; Garcia-Ranea, J.A.; Valencia, A.; Santisteban, P.; Crespo, P.; Bernal, J. The small GTP-binding protein, Rhes, regulates signal transduction from G protein-coupled receptors. Oncogene 2004, 23, 559–568.
- Hill, C.; Goddard, A.; Ladds, G.; Davey, J. The cationic region of Rhes mediates its interactions with specific Gbeta subunits. Cell Physiol. Biochem. 2009, 23, 1–8.
- Ehrenberg, A.J.; Leng, K.; Letourneau, K.N.; Hernandez, I.; Lew, C.; Seeley, W.W.; Spina, S.; Miller, B.; Heinsen, H.; Kampmann, M.; et al. Patterns of neuronal Rhes as a novel hallmark of tauopathies. Acta Neuropathol. 2021, 141, 651–666.
- Falk, J.D.; Vargiu, P.; Foye, P.E.; Usui, H.; Perez, J.; Danielson, P.E.; Lerner, D.L.; Bernal, J.; Sutcliffe, J.G. Rhes: A striatal-specific Ras homolog related to Dexras1. J. Neurosci. Res. 1999, 57, 782–788.
- Hernandez, I.; Luna, G.; Rauch, J.N.; Reis, S.A.; Giroux, M.; Karch, C.M.; Boctor, D.; Sibih, Y.E.; Storm, N.J.; Diaz, A.; et al. A farnesyltransferase inhibitor activates lysosomes and reduces tau pathology in mice with tauopathy. Sci. Transl. Med. 2019, 11, eaat3005.
- Errico, F.; Santini, E.; Migliarini, S.; Borgkvist, A.; Centonze, D.; Nasti, V.; Carta, M.; De Chiara, V.; Prosperetti, C.; Spano, D.; et al. The GTP-binding protein Rhes modulates dopamine signalling in striatal medium spiny neurons. Mol. Cell. Neurosci. 2008, 37, 335–345.
- Ghiglieri, V.; Napolitano, F.; Pelosi, B.; Schepisi, C.; Migliarini, S.; Di Maio, A.; Pendolino, V.; Mancini, M.; Sciamanna, G.; Vitucci, D.; et al. Rhes influences striatal cAMP/PKA-dependent signaling and synaptic plasticity in a gender-sensitive fashion. Sci. Rep. 2015, 5, 1–17.
- Sciamanna, G.; Napolitano, F.; Pelosi, B.; Bonsi, P.; Vitucci, D.; Nuzzo, T.; Punzo, D.; Ghiglieri, V.; Ponterio, G.; Pasqualetti, M.; et al. Rhes regulates dopamine D2 receptor transmission in striatal cholinergic interneurons. Neurobiol. Dis. 2015, 78, 146–161.
- Harrison, L.; LaHoste, G. Rhes, the Ras homolog enriched in striatum, is reduced under conditions of dopamine supersensitivity. Neuroscience 2006, 137, 483–492.
- Harrison, L.M.; LaHoste, G.J.; Ruskin, D.N. Ontogeny and dopaminergic regulation in brain of Ras homolog enriched in striatum (Rhes). Brain Res. 2008, 1245, 16–25.
- Szele, F.G.; Artymyshyn, R.; Molinoff, P.B.; Chesselet, M.-F. Heterogeneous distribution of dopamine D2 receptor mRNA in the rat striatum: A quantitative analysis with in situ hybridization histochemistry. Anat. Rec. 1991, 231, 548–558.
- Pinna, A.; Napolitano, F.; Pelosi, B.; Di Maio, A.; Wardas, J.; Casu, M.A.; Costa, G.; Migliarini, S.; Calabresi, P.; Pasqualetti, M.; et al. The small GTP-binding protein Rhes influences nigrostriatal-dependent motor behavior during aging. Mov. Disord. 2016, 31, 583–589.
- Vitucci, D.; Di Giorgio, A.; Napolitano, F.; Pelosi, B.; Blasi, G.; Errico, F.; Attrotto, M.T.; Gelao, B.; Fazio, L.; Taurisano, P.; et al. Rasd2 modulates prefronto-striatal phenotypes in humans and “Schizophrenia-like behaviors” in mice. Neuropsychopharmacology 2016, 41, 916–927.
- Manzano, J.; Morte, B.; Scanlan, T.S.; Bernal, J. Differential effects of triiodothyronine and the thyroid hormone receptor beta-specific agonist GC-1 on thyroid hormone target genes in the b ain. Endocrinology 2003, 144, 5480–5487.
- Vallortigara, J.; Alfos, S.; Micheau, J.; Higueret, P.; Enderlin, V. T3 administration in adult hypothyroid mice modulates expression of proteins involved in striatal synaptic plasticity and improves motor behavior. Neurobiol. Dis. 2008, 31, 378–385.
- Vargiu, P.; Morte, B.; Manzano, J.; Perez, J.; De Abajo, R.; Sutcliffe, J.G.; Bernal, J. Thyroid hormone regulation of rhes, a novel Ras homolog gene expressed in the striatum. Brain Res. Mol. Brain Res. 2001, 94, 1–8.
- Napolitano, F.; Warren, E.B.; Migliarini, S.; Punzo, D.; Errico, F.; Li, Q.; Thiolat, M.-L.; Vescovi, A.L.; Calabresi, P.; Bezard, E.; et al. Decreased Rhes mRNA levels in the brain of patients with Parkinson’s disease and MPTP-treated macaques. PLoS ONE 2017, 12, e0181677.
- Harrison, L.M.; He, Y. Rhes and AGS1/Dexras1 affect signaling by dopamine D1 receptors through adenylyl cyclase. J. Neurosci. Res. 2011, 89, 874–882.
- Thapliyal, A.; Bannister, R.A.; Hanks, C.; Adams, B.A. The monomeric G proteins AGS1 and Rhes selectively influence Galphai-dependent signaling to modulate N-type (CaV2.2) calcium channels. Am. J. Physiol. Cell Physiol. 2008, 295, C1417–C1426.
- Shahani, N.; Swarnkar, S.; Giovinazzo, V.; Morgenweck, J.; Bohn, L.M.; Scharager-Tapia, C.; Pascal, B.; Martinez-Acedo, P.; Khare, K.; Subramaniam, S. RasGRP1 promotes amphetamine-induced motor behavior through a Rhes interaction network (“Rhesactome”) in the striatum. Sci. Signal. 2016, 9, ra111.
- Napolitano, F.; De Rosa, A.; Russo, R.; Di Maio, A.; Garofalo, M.; Federici, M.; Migliarini, S.; LeDonne, A.; Rizzo, F.R.; Avallone, L.; et al. The striatal-enriched protein Rhes is a critical modulator of cocaine-induced molecular and behavioral responses. Sci. Rep. 2019, 9, 15294.
- Napolitano, F.; D’Angelo, L.; de Girolamo, P.; Avallone, L.; de Lange, P.; Usiello, A. The thyroid hormone-target gene Rhes a novel crossroad for neurological and psychiatric disorders: New insights from animal models. Neuroscience 2018, 384, 419–428.
- Bang, S.; Steenstra, C.; Kim, S.F. Striatum specific protein, Rhes regulates AKT pathway. Neurosci. Lett. 2012, 521, 142–147.
- Harrison, L.; Muller, S.; Spano, D. Effects of the Ras homolog Rhes on Akt/protein kinase B and glycogen synthase kinase 3 phosphorylation in striatum. Neuroscience 2013, 236, 21–30.
- Beaulieu, J.-M.; Gainetdinov, R.; Caron, M.G. The Akt-GSK-3 signaling cascade in the actions of dopamine. Trends Pharmacol. Sci. 2007, 28, 166–172.
- Wilkinson, K.A.; Henley, J.M. Mechanisms, regulation and consequences of protein SUMOylation. Biochem. J. 2010, 428, 133–145.
- Anderson, D.B.; Zanella, C.A.; Henley, J.M.; Cimarosti, H. Sumoylation: Implications for Neurodegenerative Diseases. Adv. Exp. Med. Biol. 2017, 963, 261–281.
- Subramaniam, S.; Napolitano, F.; Mealer, R.G.; Kim, S.; Errico, F.; Barrow, R.; Shahani, N.; Tyagi, R.; Snyder, S.H.; Usiello, A. Rhes, a striatal-enriched small G protein, mediates mTOR signaling and L-DOPA-induced dyskinesia. Nat. Neurosci. 2011, 15, 191–193.
- Subramaniam, S.; Sixt, K.M.; Barrow, R.; Snyder, S.H. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science 2009, 324, 1327–1330.
- Lu, B.; Palacino, J. A novel human embryonic stem cell-derived Huntington’s disease neuronal model exhibits mutant huntingtin (mHTT) aggregates and soluble mHTT-dependent neurodegeneration. FASEB J. 2013, 27, 1820–1829.
- Subramaniam, S.; Mealer, R.G.; Sixt, K.M.; Barrow, R.K.; Usiello, A.; Snyder, S.H. Rhes, a physiologic regulator of sumoylation, enhances cross-sumoylation between the basic sumoylation enzymes E1 and Ubc9. J. Biol. Chem. 2010, 285, 20428–20432.
- Mealer, R.G.; Subramaniam, S.; Snyder, S.H. Rhes deletion is neuroprotective in the 3-nitropropionic acid model of Huntington’s disease. J. Neurosci. 2013, 33, 4206–4210.
- Baiamonte, B.A.; Lee, F.A.; Brewer, S.T.; Spano, D.; LaHoste, G.J. Attenuation of Rhes activity significantly delays the appearance of behavioral symptoms in a mouse model of Huntington’s disease. PLoS ONE 2013, 8, e53606.
- Swarnkar, S.; Chen, Y.; Pryor, W.M.; Shahani, N.; Page, D.T.; Subramaniam, S. Ectopic expression of the striatal-enriched GTPase Rhes elicits cerebellar degeneration and an ataxia phenotype in Huntington’s disease. Neurobiol. Dis. 2015, 82, 66–77.
- Liu, Q.; Cheng, S.; Yang, H.; Zhu, L.; Pan, Y.; Jing, L.; Tang, B.; Li, S.; Li, X.-J. Loss of Hap1 selectively promotes striatal degeneration in Huntington disease mice. Proc. Natl. Acad. Sci. USA 2020, 117, 20265–20273.
- Sbodio, J.I.; Paul, B.D.; Machamer, C.E.; Snyder, S.H. Golgi protein ACBD3 mediates neurotoxicity associated with Huntington’s disease. Cell Rep. 2013, 4, 890–897.
- Sharma, M.; Subramaniam, S. Rhes travels from cell to cell and transports Huntington disease protein via TNT-like protrusion. J. Cell. Biol. 2019, 218, 1972–1993.
- Decressac, M.; Bjorklund, A. mTOR inhibition alleviates L-DOPA-induced dyskinesia in parkinsonian rats. J. Parkinsons Dis. 2013, 3, 13–17.
- Santini, E.; Heiman, M.; Greengard, P.; Valjent, E.; Fisone, G. Inhibition of mTOR signaling in Parkinson’s disease prevents L-DOPA-induced dyskinesia. Sci. Signal. 2009, 2, ra36.
- Obeso, J.A.; Stamelou, M.; Goetz, C.G.; Poewe, W.; Lang, A.E.; Weintraub, D.; Burn, D.; Halliday, G.M.; Bezard, E.; Przedborski, S.; et al. Past, present, and future of Parkinson’s disease: A special essay on the 200th anniversary of the Shaking Palsy. Mov. Disord. 2017, 32, 1264–1310.
- Calabresi, P.; Di Filippo, M.; Ghiglieri, V.; Tambasco, N.; Picconi, B. Levodopa-induced dyskinesias in patients with Parkinson’s disease: Filling the bench-to-bedside gap. Lancet Neurol. 2010, 9, 1106–1117.
- Cenci, M.A. Dopamine dysregulation of movement control in L-DOPA-induced dyskinesia. Trends Neurosci. 2007, 30, 236–243.
- Olanow, C.W.; Stern, M.B.; Sethi, K. The scientific and clinical basis for the treatment of Parkinson disease. Neurology 2009, 72, S1–S136.
- Picconi, B.; Centonze, D.; Håkansson, K.; Bernardi, G.; Greengard, P.; Fisone, G.; Cenci, M.A.; Calabresi, P. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat. Neurosci. 2003, 6, 501–506.
- Jenner, P. Molecular mechanisms of L-DOPA-induced dyskinesia. Nat. Rev. Neurosci. 2008, 9, 665–677.
- Subramaniam, S.; Snyder, S.H. Huntington’s disease is a disorder of the corpus striatum: Focus on Rhes (Ras homologue enriched in the striatum). Neuropharmacology 2011, 60, 1187–1192.
- Brugnoli, A.; Napolitano, F.; Usiello, A.; Morari, M. Genetic deletion of Rhes or pharmacological blockade of mTORC1 prevent striato-nigral neurons activation in levodopa-induced dyskinesia. Neurobiol. Dis. 2016, 85, 155–163.
- Eshraghi, M.; Ramírez-Jarquín, U.N.; Shahani, N.; Nuzzo, T.; De Rosa, A.; Swarnkar, S.; Galli, N.; Rivera, O.; Tsaprailis, G.; Scharager-Tapia, C.; et al. RasGRP1 is a causal factor in the development of l-DOPA-induced dyskinesia in Parkinson’s disease. Sci. Adv. 2020, 6, eaaz7001.
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840.
- Greenamyre, J.T.; Hastings, T.G. Biomedicine. Parkinson’s–Divergent causes, convergent mechanisms. Science 2004, 304, 1120–1122.
- Schapira, A.H.; Jenner, P. Etiology and pathogenesis of Parkinson’s disease. Mov. Disord. 2011, 26, 1049–1055.
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888.
- Dhandapani, K.M.; Brann, D.W. Protective effects of estrogen and selective estrogen receptor modulators in the brain. Biol. Reprod. 2002, 67, 1379–1385.
- Gillies, G.E.; Pienaar, I.S.; Vohra, S.; Qamhawi, Z. Sex differences in Parkinson’s disease. Front. Neuroendocrinol. 2014, 35, 370–384.
- Bartels, T.; De Schepper, S.; Hong, S. Microglia modulate neurodegeneration in Alzheimer’s and Parkinson’s diseases. Science 2020, 370, 66–69.
- Henry, V.; Paillé, V.; Lelan, F.; Brachet, P.; Damier, P. Kinetics of microglial activation and degeneration of dopamine-containing neurons in a rat model of Parkinson disease induced by 6-hydroxydopamine. J. Neuropathol. Exp. Neurol. 2009, 68, 1092–1102.
- Wu, D.C.; Jackson-Lewis, V.; Vila, M.; Tieu, K.; Teismann, P.; Vadseth, C.; Choi, D.-K.; Ischiropoulos, H.; Przedborski, S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J. Neurosci. 2002, 22, 1763–1771.
- Costa, G.; Pinna, A.; Porceddu, P.F.; Casu, M.A.; Di Maio, A.; Napolitano, F.; Usiello, A.; Morelli, M. Rhes counteracts dopamine neuron degeneration and neuroinflammation depending on gender and age. Front. Aging Neurosci. 2018, 10, 163.
- Barrett, S.P.; Darredeau, C.; Pihl, R.O. Patterns of simultaneous polysubstance use in drug using university students. Hum. Psychopharmacol. 2006, 21, 255–263.
- Simola, N.; Costa, G.; De Luca, M.A.; Piras, G.; Marongiu, J.; Fattore, L. Neuronal and peripheral damages induced by synthetic psychoactive substances: An update of recent findings from human and animal studies. Neural Regen. Res. 2020, 15, 802–816.
- Strote, J.; Lee, J.E.; Wechsler, H. Increasing MDMA use among college students: Results of a national survey. J. Adolesc. Health 2002, 30, 64–72.
- Baumann, M.H.; Wang, X.; Rothman, R.B. 3,4-Methylenedioxymethamphetamine (MDMA) neurotoxicity in rats: A reappraisal of past and present findings. Psychopharmacology 2007, 189, 407–424.
- Easton, N.; Marsden, C.A. Ecstasy: Are animal data consistent between species and can they translate to humans? J. Psychopharmacol. 2006, 20, 194–210.
- Gudelsky, G.A.; Yamamoto, B.K. Actions of 3,4-methylenedioxymethamphetamine (MDMA) on cerebral dopaminergic, serotonergic and cholinergic neurons. Pharmacol. Biochem. Behav. 2008, 90, 198–207.
- Parrott, A.C. Human psychopharmacology of Ecstasy (MDMA): A review of 15 years of empirical research. Hum. Psychopharmacol. 2001, 16, 557–577.
- Costa, G.; Morelli, M.; Simola, N. Progression and persistence of neurotoxicity induced by MDMA in dopaminergic regions of the mouse brain and association with noradrenergic, GABAergic, and serotonergic damage. Neurotox. Res. 2017, 32, 563–574.
- Frau, L.; Simola, N.; Porceddu, P.F.; Morelli, M. Effect of crowding, temperature and age on glia activation and dopaminergic neurotoxicity induced by MDMA in the mouse brain. Neurotoxicology 2016, 56, 127–138.
- Granado, N.; O′shea, E.; Bove, J.; Vila, M.; Colado, M.I.; Moratalla, R. Persistent MDMA-induced dopaminergic neurotoxicity in the striatum and substantia nigra of mice. J. Neurochem. 2008, 107, 1102–1112.
- Moratalla, R.; Khairnar, A.; Simola, N.; Granado, N.; García-Montes, J.R.; Porceddu, P.F.; Tizabi, Y.; Costa, G.; Morelli, M. Amphetamine-related drugs neurotoxicity in humans and in experimental animals: Main mechanisms. Prog. Neurobiol. 2017, 155, 149–170.
- Costa, G.; Porceddu, P.F.; Serra, M.; Casu, M.A.; Schiano, V.; Napolitano, F.; Pinna, A.; Usiello, A.; Morelli, M. Lack of Rhes increases MDMA-induced neuroinflammation and dopamine neuron degeneration: Role of gender and age. Int. J. Mol. Sci. 2019, 20, 1556.
More
Information
Subjects:
Neurosciences
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
859
Revisions:
2 times
(View History)
Update Date:
08 Jun 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No