 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rungroch Sungthong | + 1045 word(s) | 1045 | 2021-06-04 11:39:37 | | | |
| 2 | Enzi Gong | Meta information modification | 1045 | 2021-06-07 11:28:36 | | | | |
| 3 | Rungroch Sungthong | + 4125 word(s) | 5170 | 2021-06-07 22:37:30 | | | | |
| 4 | Enzi Gong | Meta information modification | 5170 | 2021-06-29 03:38:06 | | |
Video Upload Options
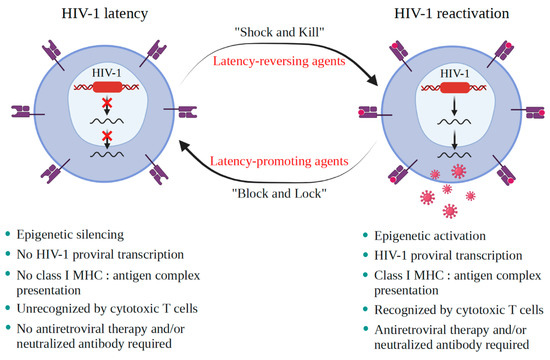
A critical burden of AIDS therapy is the evasive nature of HIV-1 in face of host immune responses, the so-called “latency.” Recently, a promising approach, the “Shock and Kill” strategy, was proposed to eliminate latently HIV-1-infected cell reservoirs. This therapeutic concept involves two crucial steps: HIV-1 reactivation from its latency stage using a latency-reversing agent (LRA) followed by host immune responses to destroy HIV-1-infected cells in combination with reinforced antiretroviral therapy to kill the progeny virus. Looking at epigenetics of HIV-1 infection, researchers proved that some bromodomains and extra-terminal motif protein inhibitors (BETis) can reactivate HIV-1 from latency. However, to date, only a few BETis have shown HIV-1-reactivating functions, and none of them have yet been approved for clinical trial.
1. Introduction
AIDS first emerged decades ago; however, its cure, i.e., eliminating all virus sources, is still unachievable. Although antiretroviral therapy in AIDS patients reduces viremia, continuous administration of drugs is required, since HIV-1 gene transcription still occurs at a residual level in latently HIV-1-infected cell reservoirs, mainly in resting CD4+ T cells, in patients under combination antiretroviral therapy (cART) [1]. This residual transcription is associated with chronic immune activation and low-level inflammation, which support non-AIDS co-morbidities [2]. However, there is now compelling evidence that the composition of HIV-1 reservoir is heterogeneous [3]. Indeed, HIV-1 is latently established in various cell types such as hematopoietic stem cells, dendritic cells, microglial cells or in cells from the monocyte-macrophage lineage reviewed in [1][2][6]. Moreover, these cells localize in a variety of anatomical sites including tissues such as the blood, the brain, the gut-associated lymphoid tissue, the adipose tissue, the bone marrow, and the genital tract [7], making it difficult to clear all virus reservoirs.
2. State of the Art of the “Shock” Step
3. Roles of BET Family Proteins and Their Inhibitors in HIV-1-Infected Cells
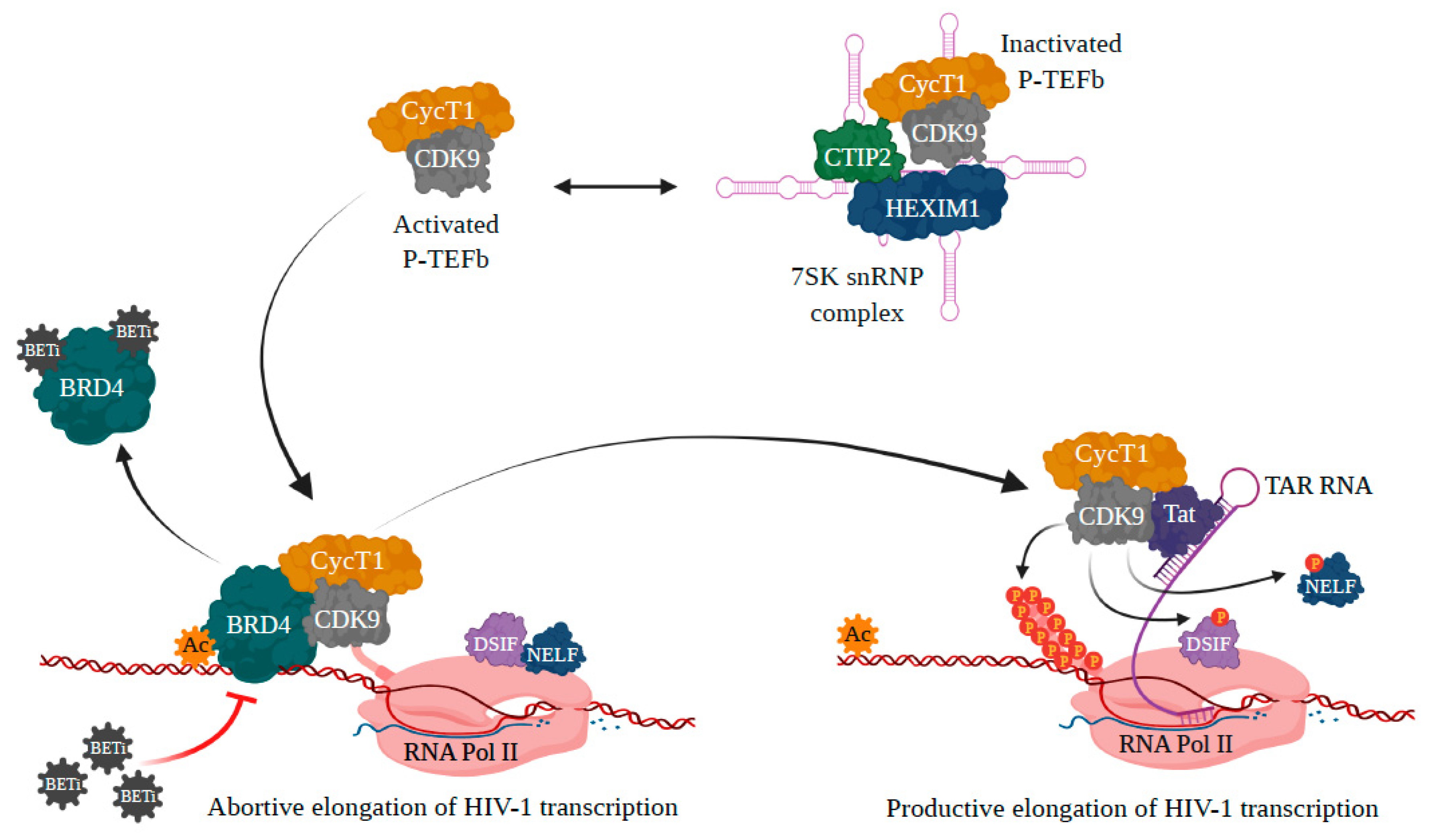
3.1. BRD4
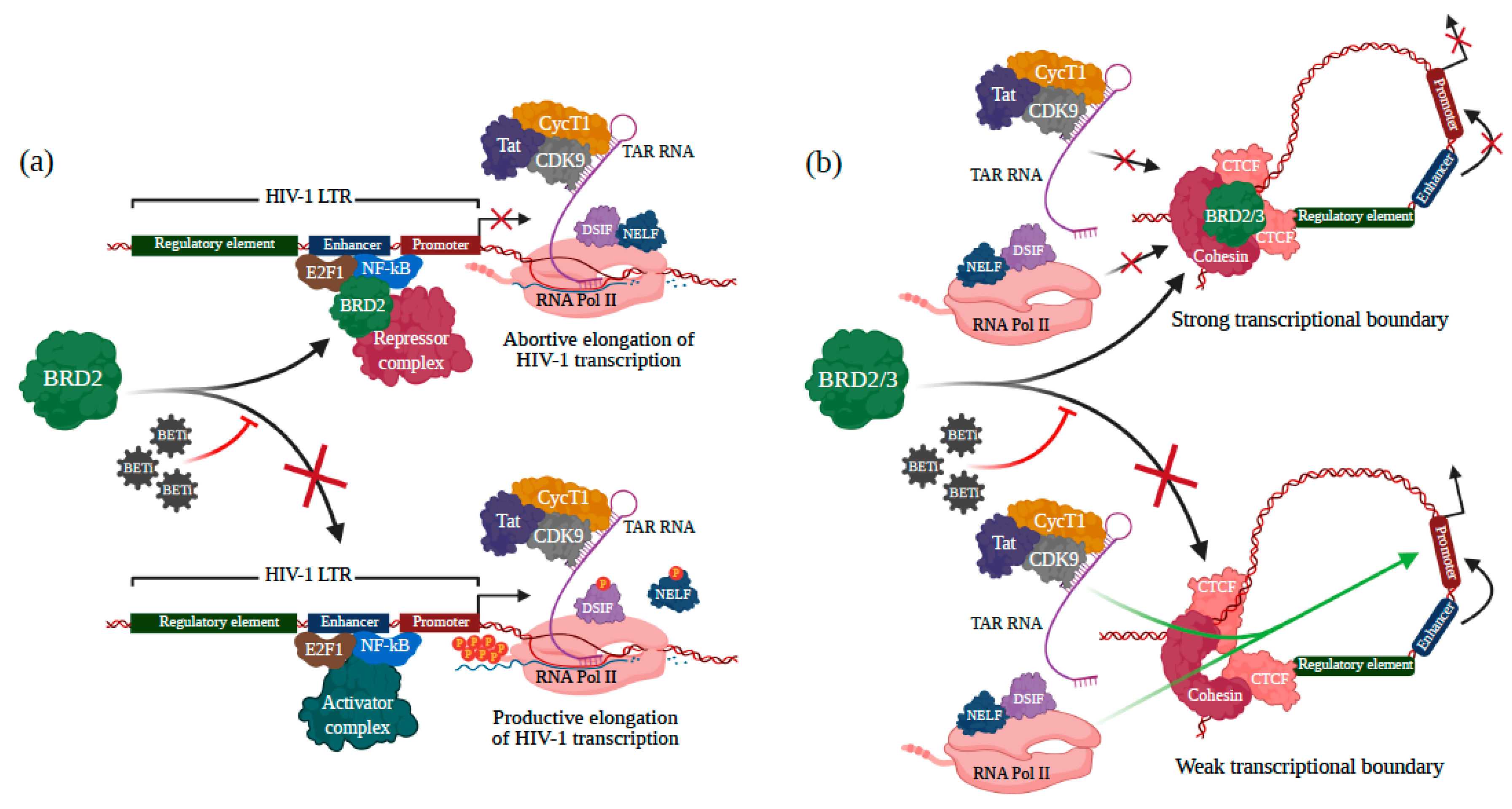
3.2. BRD2
3.3. Other BET Family Proteins
4. BETis and Immunity
4.1. Innate Immunity
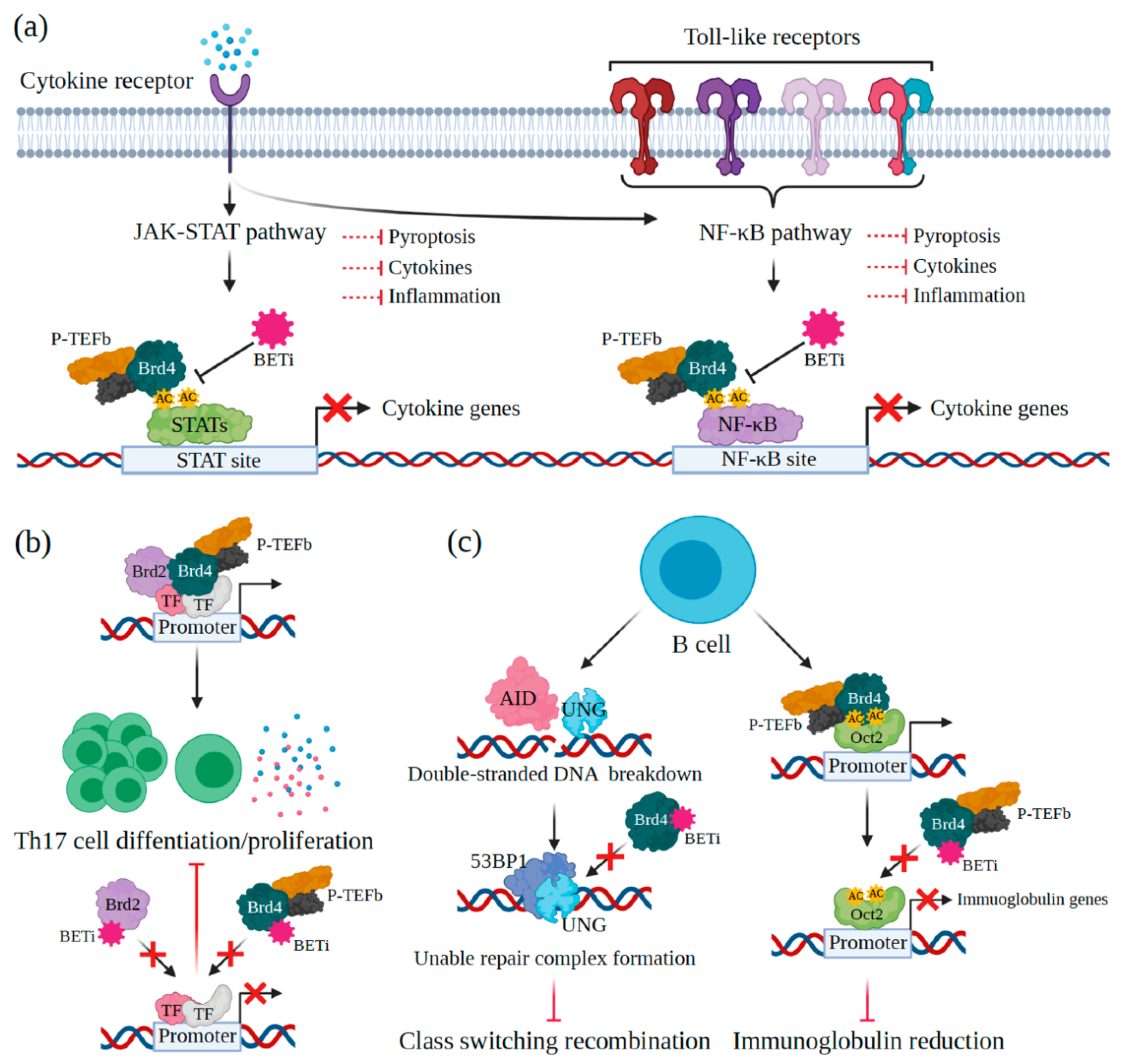
4.2. Adaptive Immunity
5. BETis: Current and Beyond
6. Conclusions
References
- Arts, E.J.; Hazuda, D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161.
- Massanella, M.; Fromentin, R.; Chomont, N. Residual inflammation and viral reservoirs: Alliance against an HIV cure. Curr. Opin. HIV AIDS 2016, 11, 234.
- Ait-Ammar, A.; Kula, A.; Darcis, G.; Verdikt, R.; de Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Rohr, O.; van Lint, C. Current status of latency reversing agents facing the heterogeneity of HIV-1 cellular and tissue reservoirs. Front. Microbiol. 2020, 10, 3060.
- Marcello, A. Latency: The hidden HIV-1 challenge. Retrovirology 2006, 3, 7.
- García, M.; Buzón, M.J.; Benito, J.M.; Rallón, N. Peering into the HIV reservoir. Rev. Med. Virol. 2018, 28, e1981.
- Sung, J.M.; Margolis, D.M. HIV persistence on antiretroviral therapy and barriers to a cure. Adv. Exp. Med. Biol. 2018, 1075, 165–185.
- Eisele, E.; Siliciano, R.F. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity 2012, 37, 377–388.
- Le Douce, V.; Ait-Amar, A.; Forouzanfar, F.; Fahmi, F.; Quiel, J.; El Mekdad, H.; Daouad, F.; Marban, C.; Rohr, O.; Schwartz, C. Improving combination antiretroviral therapy by targeting HIV-1 gene transcription. Expert Opin. Ther. Targets 2016, 20, 1311–1324.
- Le Douce, V.; Cherrier, T.; Riclet, R.; Rohr, O.; Schwartz, C. The many lives of CTIP2: From AIDS to cancer and cardiac hypertrophy. J. Cell. Physiol. 2014, 229, 533–537.
- Le Douce, V.; Forouzanfar, F.; Eilebrecht, S.; van Driessche, B.; Ait-Ammar, A.; Verdikt, R.; Kurashige, Y.; Marban, C.; Gautier, V.; Candolfi, E.; et al. HIC1 controls cellular- and HIV-1-gene transcription via interactions with CTIP2 and HMGA1. Sci. Rep. 2016, 6, 1–14.
- Marban, C.; Forouzanfar, F.; Ait-Ammar, A.; Fahmi, F.; El Mekdad, H.; Daouad, F.; Rohr, O.; Schwartz, C. Targeting the brain reservoirs: Toward an HIV cure. Front. Immunol. 2016, 7, 397.
- Marban, C.; Suzanne, S.; Dequiedt, F.; de Walque, S.; Redel, L.; van Lint, C.; Aunis, D.; Rohr, O. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J. 2007, 26, 412–423.
- Cherrier, T.; Suzanne, S.; Redel, L.; Calao, M.; Marban, C.; Samah, B.; Mukerjee, R.; Schwartz, C.; Gras, G.; Sawaya, B.E.; et al. p21WAF1 gene promoter is epigenetically silenced by CTIP2 and SUV39H1. Oncogene 2009, 28, 3380–3389.
- Le Douce, V.; Colin, L.; Redel, L.; Cherrier, T.; Herbein, G.; Aunis, D.; Rohr, O.; van Lint, C.; Schwartz, C. LSD1 cooperates with CTIP2 to promote HIV-1 transcriptional silencing. Nucleic Acids Res. 2012, 40, 1904–1915.
- Cherrier, T.; Le Douce, V.; Eilebrecht, S.; Riclet, R.; Marban, C.; Dequiedt, F.; Goumon, Y.; Paillart, J.C.; Mericskay, M.; Parlakian, A.; et al. CTIP2 is a negative regulator of P-TEFb. Proc. Natl. Acad. Sci. USA 2013, 110, 12655–12660.
- Eilebrecht, S.; Le Douce, V.; Riclet, R.; Targat, B.; Hallay, H.; van Driessche, B.; Schwartz, C.; Robette, G.; van Lint, C.; Rohr, O.; et al. HMGA1 recruits CTIP2-repressed P-TEFb to the HIV-1 and cellular target promoters. Nucleic Acids Res. 2014, 42, 4962–4971.
- Darcis, G.; van Driessche, B.; van Lint, C. HIV latency: Should we shock or lock? Trends Immunol. 2017, 38, 217–228.
- Elsheikh, M.M.; Tang, Y.; Li, D.; Jiang, G. Deep latency: A new insight into a functional HIV cure. EBioMedicine 2019, 45, 624–629.
- Mousseau, G.; Aneja, R.; Clementz, M.A.; Mediouni, S.; Lima, N.S.; Haregot, A.; Kessing, C.F.; Jablonski, J.A.; Thenin-Houssier, S.; Nagarsheth, N.; et al. Resistance to the Tat inhibitor didehydro-cortistatin A is mediated by heightened basal HIV-1 transcription. mBio 2019, 10, e01750-18.
- Planas, D.; Pagliuzza, A.; Ponte, R.; Fert, A.; Marchand, L.R.; Massanella, M.; Gosselin, A.; Mehraj, V.; Dupuy, F.P.; Isnard, S.; et al. LILAC pilot study: Effects of metformin on mTOR activation and HIV reservoir persistence during antiretroviral therapy. EBioMedicine 2021, 65, 103270.
- Rohr, O. Flower power: Locking HIV in the gut with French lilac. EBioMedicine 2021, 66, 103299.
- White, C.H.; Johnston, H.E.; Moesker, B.; Manousopoulou, A.; Margolis, D.M.; Richman, D.D.; Spina, C.A.; Garbis, S.D.; Woelk, C.H.; Beliakova-Bethell, N. Mixed effects of suberoylanilide hydroxamic acid (SAHA) on the host transcriptome and proteome and their implications for HIV reactivation from latency. Antiviral. Res. 2015, 123, 78–85.
- Jeng, M.Y.; Ali, I.; Ott, M. Manipulation of the host protein acetylation network by human immunodeficiency virus type 1. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 314–325.
- Van Lint, C.; Emiliani, S.; Ott, M.; Verdin, E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996, 15, 1112–1120.
- Quivy, V.; van Lint, C. Diversity of acetylation targets and roles in transcriptional regulation: The human immunodeficiency virus type 1 promoter as a model system. Biochem. Pharmacol. 2002, 64, 925–934.
- Reuse, S.; Calao, M.; Kabeya, K.; Guiguen, A.; Gatot, J.S.; Quivy, V.; Vanhulle, C.; Lamine, A.; Vaira, D.; Demonte, D.; et al. Synergistic activation of HIV-1 expression by deacetylase inhibitors and prostratin: Implications for treatment of latent infection. PLoS ONE 2009, 4, e6093.
- Bouchat, S.; Delacourt, N.; Kula, A.; Darcis, G.; van Driessche, B.; Corazza, F.; Gatot, J.S.; Melard, A.; Vanhulle, C.; Kabeya, K.; et al. Sequential treatment with 5-aza-2’-deoxycytidine and deacetylase inhibitors reactivates HIV-1. EMBO Mol. Med. 2016, 8, 117–138.
- Bouchat, S.; Gatot, J.S.; Kabeya, K.; Cardona, C.; Colin, L.; Herbein, G.; de Wit, S.; Clumeck, N.; Lambotte, O.; Rouzioux, C.; et al. Histone methyltransferase inhibitors induce HIV-1 recovery in resting CD4+ T cells from HIV-1-infected HAART-treated patients. AIDS 2012, 26, 1473–1482.
- Subramanian, S.; Bates, S.E.; Wright, J.J.; Espinoza-Delgado, I.; Piekarz, R.L. Clinical toxicities of histone deacetylase inhibitors. Pharmaceuticals 2010, 3, 2751–2767.
- Barboric, M.; Nissen, R.M.; Kanazawa, S.; Jabrane-Ferrat, N.; Peterlin, B.M. NF-ĸB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol. Cell 2001, 8, 327–337.
- Jiang, G.; Dandekar, S. Targeting NF-κB signaling with protein kinase C agonists as an emerging strategy for combating HIV latency. AIDS Res. Hum. Retrovir. 2015, 31, 4–12.
- Rice, A.P. Cyclin-dependent kinases as therapeutic targets for HIV-1 infection. Expert Opin. Ther. Targets 2016, 20, 1453–1461.
- Sung, T.L.; Rice, A.P. Effects of prostratin on cyclin T1/P-TEFb function and the gene expression profile in primary resting CD4+ T cells. Retrovirology 2006, 3, 66.
- Fujinaga, K.; Barboric, M.; Li, Q.; Luo, Z.; Price, D.H.; Peterlin, B.M. PKC phosphorylates HEXIM1 and regulates P-TEFb activity. Nucleic Acids Res. 2012, 40, 9160–9170.
- Pandeló José, D.; Bartholomeeusen, K.; da Cunha, R.D.; Abreu, C.M.; Glinski, J.; da Costa, T.B.; Bacchi Rabay, A.F.; Pianowski Filho, L.F.; Dudycz, L.W.; Ranga, U.; et al. Reactivation of latent HIV-1 by new semi-synthetic ingenol esters. Virology 2014, 462–463, 328–339.
- Darcis, G.; Kula, A.; Bouchat, S.; Fujinaga, K.; Corazza, F.; Ait-Ammar, A.; Delacourt, N.; Melard, A.; Kabeya, K.; Vanhulle, C.; et al. An in-depth comparison of latency-reversing agent combinations in various in vitro and ex vivo HIV-1 latency models identified bryostatin-1+JQ1 and ingenol-B+JQ1 to potently reactivate viral gene expression. PLoS Pathog. 2015, 11, e1005063.
- Abner, E.; Jordan, A. HIV “shock and kill” therapy: In need of revision. Antiviral Res. 2019, 166, 19–34.
- Cary, D.C.; Fujinaga, K.; Peterlin, B.M. Molecular mechanisms of HIV latency. J. Clin. Investig. 2016, 126, 448–454.
- Darcis, G.; van Driessche, B.; van Lint, C. Preclinical shock strategies to reactivate latent HIV-1: An update. Curr. Opin. HIV AIDS 2016, 11, 388–393.
- van Lint, C.; Bouchat, S.; Marcello, A. HIV-1 transcription and latency: An update. Retrovirology 2013, 10, 67.
- Ait-Ammar, A.; Bellefroid, M.; Daouad, F.; Martinelli, V.; van Assche, J.; Wallet, C.; Rodari, A.; de Rovere, M.; Fahrenkrog, B.; Schwartz, C.; et al. Inhibition of HIV-1 gene transcription by KAP1 in myeloid lineage. Sci. Rep. 2021, 11, 2692.
- Deeks, S.G.; Lewin, S.R.; Ross, A.L.; Ananworanich, J.; Benkirane, M.; Cannon, P.; Chomont, N.; Douek, D.; Lifson, J.D.; Lo, Y.R.; et al. International AIDS Society global scientific strategy: Towards an HIV cure 2016. Nat. Med. 2016, 22, 839–850.
- Le Douce, V.; Janossy, A.; Hallay, H.; Ali, S.; Riclet, R.; Rohr, O.; Schwartz, C. Achieving a cure for HIV infection: Do we have reasons to be optimistic? J. Antimicrob. Chemother. 2012, 67, 1063–1074.
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073.
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a syn-thetic histone mimic. Nature 2010, 468, 1119–1123.
- Padmanabhan, B.; Mathur, S.; Manjula, R.; Tripathi, S. Bromodomain and extra-terminal (BET) family proteins: New thera-peutic targets in major diseases. J. Biosci. 2016, 41, 295–311.
- Banerjee, C.; Archin, N.; Michaels, D.; Belkina, A.C.; Denis, G.V.; Bradner, J.; Sebastiani, P.; Margolis, D.M.; Montano, M. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J. Leukoc. Biol. 2012, 92, 1147–1154.
- Zhu, J.; Gaiha, G.D.; John, S.P.; Pertel, T.; Chin, C.R.; Gao, G.; Qu, H.; Walker, B.D.; Elledge, S.J.; Brass, A.L. Reactivation of latent HIV-1 by inhibition of BRD4. Cell Rep. 2012, 2, 807–816.
- Yang, A.Y.; Kim, H.; Li, W.; Kong, A.N. Natural compound-derived epigenetic regulators targeting epigenetic readers, writers and erasers. Curr. Top. Med. Chem. 2016, 16, 697–713.
- Huang, B.; Li, G.; Jiang, X.H. Fate determination in mesenchymal stem cells: A perspective from histone-modifying enzymes. Stem Cell Res. Ther. 2015, 6, 35.
- Lusic, M.; Siliciano, R.F. Nuclear landscape of HIV-1 infection and integration. Nat. Rev. Microbiol. 2017, 15, 69–82.
- Lange, U.C.; Verdikt, R.; Ait-Ammar, A.; van Lint, C. Epigenetic crosstalk in chronic infection with HIV-1. Semin. Immunopathol. 2020, 42, 187–200.
- Du Chéné, I.; Basyuk, E.; Lin, Y.L.; Triboulet, R.; Knezevich, A.; Chable-Bessia, C.; Mettling, C.; Baillat, V.; Reynes, J.; Corbeau, P.; et al. Suv39H1 and HP1γ are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J. 2007, 26, 424–435.
- Taniguchi, Y. The bromodomain and extra-terminal domain (BET) family: Functional anatomy of BET paralogous proteins. Int. J. Mol. Sci. 2016, 17, 1849.
- Sartor, G.C.; Malvezzi, A.M.; Kumar, A.; Andrade, N.S.; Wiedner, H.J.; Vilca, S.J.; Janczura, K.J.; Bagheri, A.; Al-Ali, H.; Powell, S.K.; et al. Enhancement of BDNF expression and memory by HDAC inhibition requires BET bromodomain reader proteins. J. Neurosci. 2019, 39, 612–626.
- Boehm, D.; Conrad, R.J.; Ott, M. Bromodomain proteins in HIV infection. Viruses 2013, 5, 1571–1586.
- Huang, B.; Yang, X.D.; Zhou, M.M.; Ozato, K.; Chen, L.F. Brd4 coactivates transcriptional activation of NF-ĸB via specific binding to acetylated RelA. Mol. Cell. Biol. 2009, 29, 1375–1387.
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545.
- Bisgrove, D.A.; Mahmoudi, T.; Henklein, P.; Verdin, E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 13690–13695.
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.S.; Brady, J.N.; Ozato, K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 2005, 19, 523–534.
- Yang, Z.; Zhu, Q.; Luo, K.; Zhou, Q. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature 2001, 414, 317–322.
- Schröder, S.; Cho, S.; Zeng, L.; Zhang, Q.; Kaehlcke, K.; Mak, L.; Lau, J.; Bisgrove, D.; Schnölzer, M.; Verdin, E.; et al. Two-pronged binding with bromodomain-containing protein 4 liberates positive transcription elongation factor b from inactive ribonucleoprotein complexes. J. Biol. Chem. 2012, 287, 1090–1099.
- Li, Z.; Guo, J.; Wu, Y.; Zhou, Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013, 41, 277–287.
- Zhou, M.; Huang, K.; Jung, K.J.; Cho, W.K.; Klase, Z.; Kashanchi, F.; Pise-Masison, C.A.; Brady, J.N. Bromodomain protein Brd4 regulates human immunodeficiency virus transcription through phosphorylation of CDK9 at threonine 29. J. Virol. 2009, 83, 1036–1044.
- Wallet, C.; Rohr, O.; Schwartz, C. Evolution of a concept: From accessory protein to key virulence factor, the case of HIV-1 Vpr. Biochem. Pharmacol. 2020, 180, 114128.
- Wang, J.; Reuschel, E.L.; Shackelford, J.M.; Jeang, L.; Shivers, D.K.; Diehl, J.A.; Yu, X.-F.; Finkel, T.H. HIV-1 Vif promotes the G1-to S-phase cell-cycle transition. Blood 2011, 117, 1260–1269.
- Boehm, D.; Calvanese, V.; Dar, R.D.; Xing, S.; Schroeder, S.; Martins, L.; Aull, K.; Li, P.C.; Planelles, V.; Bradner, J.E.; et al. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle 2013, 12, 452–462.
- Conrad, R.J.; Fozouni, P.; Thomas, S.; Sy, H.; Zhang, Q.; Zhou, M.M.; Ott, M. The short isoform of BRD4 promotes HIV-1 latency by engaging repressive SWI/SNF chromatin-remodeling complexes. Mol. Cell 2017, 67, 1001–1012.
- Alamer, E.; Zhong, C.; Hajnik, R.; Soong, L.; Hu, H. Modulation of BRD4 in HIV epigenetic regulation: Implications for finding an HIV cure. Retrovirology 2021, 18, 3.
- Lu, P.; Qu, X.; Shen, Y.; Jiang, Z.; Wang, P.; Zeng, H.; Ji, H.; Deng, J.; Yang, X.; Li, X.; et al. The BET inhibitor OTX015 reac-tivates latent HIV-1 through P-TEFb. Sci. Rep. 2016, 6, 1–13.
- Beck, S.; Hanson, I.; Kelly, A.; Pappin, D.J.; Trowsdale, J. A homologue of the Drosophila female sterile homeotic (fsh) gene in the class II region of the human MHC. DNA Sequence 1992, 2, 203–210.
- Malovannaya, A.; Lanz, R.B.; Jung, S.Y.; Bulynko, Y.; Le, N.T.; Chan, D.W.; Ding, C.; Shi, Y.; Yucer, N.; Krenciute, G.; et al. Analysis of the human endogenous coregulator complexome. Cell 2011, 145, 787–799.
- Denis, G.V.; McComb, M.E.; Faller, D.V.; Sinha, A.; Romesser, P.B.; Costello, C.E. Identification of transcription complexes that contain the double bromodomain protein Brd2 and chromatin remodeling machines. J. Proteome Res. 2006, 5, 502–511.
- Kundu, S.K.; Katzenstein, D.; Valentine, F.T.; Spino, C.; Efron, B.; Merigan, T.C. Effect of therapeutic immunization with recombinant gp160 HIV-1 vaccine on HIV-1 proviral DNA and plasma RNA: Relationship to cellular immune responses. J. Acquir. Immune Defic. Syndr. 1997, 15, 269–274.
- Karn, J. A new BET on the control of HIV latency. Cell Cycle 2013, 12, 545–546.
- Hsu, S.C.; Gilgenast, T.G.; Bartman, C.R.; Edwards, C.R.; Stonestrom, A.J.; Huang, P.; Emerson, D.J.; Evans, P.; Werner, M.T.; Keller, C.A.; et al. The BET protein BRD2 cooperates with CTCF to enforce transcriptional and architectural boundaries. Mol. Cell 2017, 66, 102–116.
- Li, Y.; Li, G.; Ivanova, A.; Aaron, S.; Simm, M. The critical role of human transcriptional repressor CTCF mRNA up-regulation in the induction of anti-HIV-1 responses in CD4+ T cells. Immunol. Lett. 2008, 117, 35–44.
- Roberts, T.C.; Etxaniz, U.; Dall’Agnese, A.; Wu, S.Y.; Chiang, C.M.; Brennan, P.E.; Wood, M.J.A.; Puri, P.L. BRD3 and BRD4 BET bromodomain proteins differentially regulate skeletal myogenesis. Sci. Rep. 2017, 7, 1–16.
- LeRoy, G.; Rickards, B.; Flint, S.J. The double bromodomain proteins Brd2 and Brd3 couple histone acetylation to transcription. Mol. Cell 2008, 30, 51–60.
- Gamsjaeger, R.; Webb, S.R.; Lamonica, J.M.; Billin, A.; Blobel, G.A.; Mackay, J.P. Structural basis and specificity of acetylated transcription factor GATA1 recognition by BET family bromodomain protein Brd3. Mol. Cell. Biol. 2011, 31, 2632–2640.
- Ren, W.; Wang, C.; Wang, Q.; Zhao, D.; Zhao, K.; Sun, D.; Liu, X.; Han, C.; Hou, J.; Li, X.; et al. Bromodomain protein Brd3 promotes Ifnb1 transcription via enhancing IRF3/p300 complex formation and recruitment to Ifnb1 promoter in macrophages. Sci. Rep. 2017, 7, 39986.
- Hsu, S.C.; Blobel, G.A. The role of bromodomain and extraterminal motif (BET) proteins in chromatin structure. Cold Spring Harb. Symp. Quant. Biol. 2017, 82, 37–43.
- Govin, J.; Lestrat, C.; Caron, C.; Pivot-Pajot, C.; Rousseaux, S.; Khochbin, S. Histone acetylation-mediated chromatin com-paction during mouse spermatogenesis. In The Histone Code and Beyond; Springer: Berlin, Germany, 2006; pp. 155–172.
- Bourova-Flin, E.; Chuffart, F.; Rousseaux, S.; Khochbin, S. The role of bromodomain testis-specific factor, BRDT, in cancer: A biomarker and a possible therapeutic target. Cell J. 2017, 19, 1–8.
- Chang, J.J.; Altfeld, M. Innate immune activation in primary HIV-1 infection. J. Infect. Dis. 2010, 202 (Suppl. 2), S297–S301.
- Iwasaki, A. Innate immune recognition of HIV-1. Immunity 2012, 37, 389–398.
- Sumner, R.P.; Thorne, L.G.; Fink, D.L.; Khan, H.; Milne, R.S.; Towers, G.J. Are evolution and the intracellular innate immune system key determinants in HIV transmission? Front. Immunol. 2017, 8, 1246.
- Wang, N.; Wu, R.; Tang, D.; Kang, R. The BET family in immunity and disease. Signal Transduct. Target. Ther. 2021, 6, 1–22.
- Wainstein, M.V.; Mossmann, M.; Araujo, G.N.; Gonçalves, S.C.; Gravina, G.L.; Sangalli, M.; Veadrigo, F.; Matte, R.; Reich, R.; Costa, F.G.; et al. Elevated serum interleukin-6 is predictive of coronary artery disease in intermediate risk overweight patients referred for coronary angiography. Diabetol. Metab. Syndr. 2017, 9, 1–7.
- Freeman, M.L.; Shive, C.L.; Nguyen, T.P.; Younes, S.A.; Panigrahi, S.; Lederman, M.M. Cytokines and T-cell homeostasis in HIV infection. J. Infect. Dis. 2016, 214 (Suppl. 2), S51–S57.
- Borges, Á.H.; O’Connor, J.L.; Phillips, A.N.; Rönsholt, F.F.; Pett, S.; Vjecha, M.J.; French, M.A.; Lundgren, J.D. Factors associated with plasma IL-6 levels during HIV infection. J. Infect. Dis. 2015, 212, 585–595.
- Keating, S.M.; Golub, E.T.; Nowicki, M.; Young, M.; Anastos, K.; Crystal, H.; Cohen, M.H.; Zhang, J.; Greenblatt, R.M.; Desai, S.; et al. The effect of HIV infection and HAART on inflammatory biomarkers in a population-based cohort of US women. AIDS 2011, 25, 1823–1832.
- Muema, D.M.; Akilimali, N.A.; Ndumnego, O.C.; Rasehlo, S.S.; Durgiah, R.; Ojwach, D.B.; Ismail, N.; Dong, K.L.; Ndhlovu, Z.M.; Mabuka, J.M.; et al. Association between the cytokine storm, immune cell dynamics, and viral replicative capacity in hyperacute HIV infection. BMC Med. 2020, 18, 1–17.
- Akase, I.E.; Obiako, R.O.; Musa, B.O.; Opawoye, A.; Akanmu, A.S. Levels of interleukin 6 and 10 and their relationship to hematological changes in HIV treatment-naïve and treatment-experienced patients. Sub-Saharan Afr. J. Med. 2019, 6, 90–95.
- Parameswaran, N.; Patial, S. Tumor necrosis factor-α signaling in macrophages. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 87–103.
- Tenorio, A.R.; Zheng, Y.; Bosch, R.J.; Krishnan, S.; Rodriguez, B.; Hunt, P.W.; Plants, J.; Seth, A.; Wilson, C.C.; Deeks, S.G.; et al. Soluble markers of inflammation and coagulation but not T-cell activation predict non–AIDS-defining morbid events during suppressive antiretroviral treatment. J. Infect. Dis. 2014, 210, 1248–1259.
- Kuller, L.H.; Tracy, R.; Belloso, W.; de Wit, S.; Drummond, F.; Lane, H.C.; Ledergerber, B.; Lundgren, J.; Neuhaus, J.; Nixon, D.; et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 2008, 5, e203.
- Maksylewicz, A.; Bysiek, A.; Lagosz, K.B.; Macina, J.M.; Kantorowicz, M.; Bereta, G.; Sochalska, M.; Gawron, K.; Chomyszyn-Gajewska, M.; Potempa, J.; et al. BET bromodomain inhibitors suppress inflammatory activation of gingival fibroblasts and epithelial cells from periodontitis patients. Front. Immunol. 2019, 10, 933.
- Nguyen, T.H.; Maltby, S.; Eyers, F.; Foster, P.S.; Yang, M. Bromodomain and extra terminal (BET) inhibitor suppresses macrophage-driven steroid-resistant exacerbations of airway hyper-responsiveness and inflammation. PLoS ONE 2016, 11, e0163392.
- Jahagirdar, R.; Zhang, H.; Azhar, S.; Tobin, J.; Attwell, S.; Yu, R.; Wu, J.; McLure, K.G.; Hansen, H.C.; Wagner, G.S.; et al. A novel BET bromodomain inhibitor, RVX-208, shows reduction of atherosclerosis in hyperlipidemic ApoE deficient mice. Atherosclerosis 2014, 236, 91–100.
- Belkina, A.C.; Nikolajczyk, B.S.; Denis, G.V. BET protein function is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. J. Immunol. 2013, 190, 3670–3678.
- Remke, N.; Bisht, S.; Oberbeck, S.; Nolting, J.; Brossart, P. Selective BET-bromodomain inhibition by JQ1 suppresses dendritic cell maturation and antigen-specific T-cell responses. Cancer Immunol. Immunother. 2021, 70, 107–121.
- Schilderink, R.; Bell, M.; Reginato, E.; Patten, C.; Rioja, I.; Hilbers, F.W.; Kabala, P.A.; Reedquist, K.A.; Tough, D.F.; Tak, P.P.; et al. BET bromodomain inhibition reduces maturation and enhances tolerogenic properties of human and mouse dendritic cells. Mol. Immunol. 2016, 79, 66–76.
- Saravia, J.; Chapman, N.M.; Chi, H. Helper T cell differentiation. Cell. Mol. Immunol. 2019, 16, 634–643.
- Guglani, L.; Khader, S.A. Th17 cytokines in mucosal immunity and inflammation. Curr. Opin. HIV AIDS 2010, 5, 120–127.
- Gibbons, H.R.; Mi, D.J.; Farley, V.M.; Esmond, T.; Kaood, M.B.; Aune, T.M. Bromodomain inhibitor JQ1 reversibly blocks IFN-γ production. Sci. Rep. 2019, 9, 1–10.
- Watanabe, D.; Uehira, T.; Suzuki, S.; Matsumoto, E.; Ueji, T.; Hirota, K.; Minami, R.; Takahama, S.; Hayashi, K.; Sawamura, M.; et al. Clinical characteristics of HIV-1-infected patients with high levels of plasma interferon-γ: A multicenter observational study. BMC Infect. Dis. 2019, 19, 1–10.
- Roff, S.R.; Song, E.N.; Yamamoto, J.K. The significance of interferon-γ in HIV-1 pathogenesis, therapy, and prophylaxis. Front. Immunol. 2014, 4, 498.
- Nakayama, T.; Yamashita, M. Initiation and maintenance of Th2 cell identity. Curr. Opin. Immunol. 2008, 20, 265–271.
- Malhotra, P.; Haslett, P.; Sherry, B.; Shepp, D.H.; Barber, P.; Abshier, J.; Roy, U.; Schmidtmayerova, H. Increased plasma levels of the TH2 chemokine CCL18 associated with low CD4+ T cell counts in HIV-1-infected patients with a suppressed viral load. Sci. Rep. 2019, 9, 1–8.
- Planas, D.; Routy, J.P.; Ancuta, P. New Th17-specific therapeutic strategies for HIV remission. Curr. Opin. HIV AIDS 2019, 14, 85–92.
- Bixler, S.L.; Mattapallil, J.J. Loss and dysregulation of Th17 cells during HIV infection. Clin. Dev. Immunol. 2013, 2013.
- de Araújo, E.F.; Feriotti, C.; Galdino, N.A.D.L.; Preite, N.W.; Calich, V.L.G.; Loures, F.V. The iDO–ahr axis controls Th17/Treg immunity in a pulmonary model of fungal infection. Front. Immunol. 2017, 8, 880.
- Tian, C.Q.; Chen, L.; Chen, H.D.; Huan, X.J.; Hu, J.P.; Shen, J.K.; Xiong, B.; Wang, Y.Q.; Miao, Z.H. Inhibition of the BET family reduces its new target gene IDO1 expression and the production of L-kynurenine. Cell Death Dis. 2019, 10, 1–13.
- Cheung, K.L.; Zhang, F.; Jaganathan, A.; Sharma, R.; Zhang, Q.; Konuma, T.; Shen, T.; Lee, J.Y.; Ren, C.; Chen, C.H.; et al. Distinct roles of Brd2 and Brd4 in potentiating the transcriptional program for Th17 cell differentiation. Mol. Cell 2017, 65, 1068–1080.
- Ghosh, S.; Lora, J.M. Suppression of TH17-mediated pathology through BET bromodomain inhibition. Drug Discov. Today Technol. 2016, 19, 39–44.
- Mele, D.A.; Salmeron, A.; Ghosh, S.; Huang, H.R.; Bryant, B.M.; Lora, J.M. BET bromodomain inhibition suppresses TH17-mediated pathology. J. Exp. Med. 2013, 210, 2181–2190.
- Rocco, J.; Mellors, J.W.; Macatangay, B.J. Regulatory T cells: The ultimate HIV reservoir? J. Virus Erad. 2018, 4, 209–214.
- Abdel-Hameed, E.A.; Ji, H.; Sherman, K.E.; Shata, M.T.M. Epigenetic modification of FOXP3 in patients with chronic HIV infection. J. Acquir. Immune Defic. Syndr. 2014, 65, 19.
- Grant, C.; Oh, U.; Fugo, K.; Takenouchi, N.; Griffith, C.; Yao, K.; Newhook, T.E.; Ratner, L.; Jacobson, S. Foxp3 represses retroviral transcription by targeting both NF-κB and CREB pathways. PLoS Pathog. 2006, 2, e33.
- Kint, S.; Trypsteen, W.; De Spiegelaere, W.; Malatinkova, E.; Kinloch-de Loes, S.; De Meyer, T.; Van Criekinge, W.; Vandekerckhove, L. Underestimated effect of intragenic HIV-1 DNA methylation on viral transcription in infected individuals. Clin. Epigenetics 2020, 12, 1–11.
- Adeegbe, D.O.; Liu, S.; Hattersley, M.M.; Bowden, M.; Zhou, C.W.; Li, S.; Vlahos, R.; Grondine, M.; Dolgalev, I.; Ivanova, E.V.; et al. BET Bromodomain inhibition cooperates with PD-1 blockade to facilitate antitumor response in Kras-mutant non–small cell lung cancer. Cancer Immunol. Res. 2018, 6, 1234–1245.
- Collins, D.R.; Gaiha, G.D.; Walker, B.D. CD8+ T cells in HIV control, cure and prevention. Nat. Rev. Immunol. 2020, 20, 471–482.
- Gulzar, N.; Copeland, K.F. CD8+ T-cells: Function and response to HIV infection. Curr. HIV Res. 2004, 2, 23–37.
- Georgiev, P.; Wang, Y.; Muise, E.S.; Bandi, M.L.; Blumenschein, W.; Sathe, M.; Pinheiro, E.M.; Shumway, S.D. BET bromodomain inhibition suppresses human T cell function. Immunohorizons 2019, 3, 294–305.
- Gegonne, A.; Chen, Q.R.; Dey, A.; Etzensperger, R.; Tai, X.; Singer, A.; Meerzaman, D.; Ozato, K.; Singer, D.S. Immature CD8 single-positive thymocytes are a molecularly distinct subpopulation, selectively dependent on BRD4 for their differentiation. Cell Rep. 2018, 24, 117–129.
- Kagoya, Y.; Nakatsugawa, M.; Yamashita, Y.; Ochi, T.; Guo, T.; Anczurowski, M.; Saso, K.; Butler, M.O.; Arrowsmith, C.H.; Hirano, N. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J. Clin. Investig. 2016, 126, 3479–3494.
- Fenwick, C.; Joo, V.; Jacquier, P.; Noto, A.; Banga, R.; Perreau, M.; Pantaleo, G. T-cell exhaustion in HIV infection. Immunol. Rev. 2019, 292, 149–163.
- Saeidi, A.; Zandi, K.; Cheok, Y.Y.; Saeidi, H.; Wong, W.F.; Lee, C.Y.Q.; Cheong, H.C.; Yong, Y.K.; Larsson, M.; Shankar, E.M. T-cell exhaustion in chronic infections: Reversing the state of exhaustion and reinvigorating optimal protective immune responses. Front. Immunol. 2018, 9, 2569.
- Jing, X.; Shao, S.; Zhang, Y.; Luo, A.; Zhao, L.; Zhang, L.; Gu, S.; Zhao, X. BRD4 inhibition suppresses PD-L1 expression in triple-negative breast cancer. Exp. Cell Res. 2020, 392, 112034.
- Zhu, H.; Bengsch, F.; Svoronos, N.; Rutkowski, M.R.; Bitler, B.G.; Allegrezza, M.J.; Yokoyama, Y.; Kossenkov, A.V.; Bradner, J.E.; Conejo-Garcia, J.R.; et al. BET bromodomain inhibition promotes anti-tumor immunity by suppressing PD-L1 expression. Cell Rep. 2016, 16, 2829–2837.
- Moir, S.; Fauci, A.S. B-cell responses to HIV infection. Immunol. Rev. 2017, 275, 33–48.
- Zabel, F.; Mohanan, D.; Bessa, J.; Link, A.; Fettelschoss, A.; Saudan, P.; Kündig, T.M.; Bachmann, M.F. Viral particles drive rapid differentiation of memory B cells into secondary plasma cells producing increased levels of antibodies. J. Immunol. 2014, 192, 5499–5508.
- Mouquet, H. Antibody B cell responses in HIV-1 infection. Trends Immunol. 2014, 35, 549–561.
- Li, X.; Baek, G.; Ramanand, S.G.; Sharp, A.; Gao, Y.; Yuan, W.; Welti, J.; Rodrigues, D.N.; Dolling, D.; Figueiredo, I.; et al. BRD4 promotes DNA repair and mediates the formation of TMPRSS2-ERG gene rearrangements in prostate cancer. Cell Rep. 2018, 22, 796–808.
- Stanlie, A.; Yousif, A.S.; Akiyama, H.; Honjo, T.; Begum, N.A. Chromatin reader Brd4 functions in Ig class switching as a repair complex adaptor of nonhomologous end-joining. Mol. Cell 2014, 55, 97–110.
- Schormann, N.; Ricciardi, R.; Chattopadhyay, D. Uracil-DNA glycosylases—Structural and functional perspectives on an essential family of DNA repair enzymes. Protein Sci. 2014, 23, 1667–1685.
- Ward, I.M.; Minn, K.; van Deursen, J.; Chen, J. p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol. Cell. Biol. 2003, 23, 2556–2563.
- Shim, J.M.; Lee, J.S.; Russell, K.E.; Wiegman, C.H.; Barnes, P.J.; Fear, D.; Adcock, I.M.; Durham, A.L. BET proteins are a key component of immunoglobulin gene expression. Epigenomics 2017, 9, 393–406.
- Nicholls, S.J.; Schwartz, G.G.; Buhr, K.A.; Ginsberg, H.N.; Johansson, J.O.; Kalantar-Zadeh, K.; Kulikowski, E.; Toth, P.P.; Wong, N.; Sweeney, M.; et al. Apabetalone and hospitalization for heart failure in patients following an acute coronary syndrome: A prespecified analysis of the BETonMACE study. Cardiovasc. Diabetol. 2021, 20, 1–9.
- Ameratunga, M.; Braña, I.; Bono, P.; Postel-Vinay, S.; Plummer, R.; Aspegren, J.; Korjamo, T.; Snapir, A.; de Bono, J.S. First-in-human Phase 1 open label study of the BET inhibitor ODM-207 in patients with selected solid tumours. Br. J. Cancer 2020, 123, 1730–1736.
- Li, G.; Zhang, Z.; Reszka-Blanco, N.; Li, F.; Chi, L.; Ma, J.; Jeffrey, J.; Cheng, L.; Su, L. Specific activation in vivo of HIV-1 by a bromodomain inhibitor from monocytic cells in humanized mice under antiretroviral therapy. J. Virol. 2019, 93, e00233-19.
- Lu, P.; Shen, Y.; Yang, H.; Wang, Y.; Jiang, Z.; Yang, X.; Zhong, Y.; Pan, H.; Xu, J.; Lu, H.; et al. BET inhibitors RVX-208 and PFI-1 reactivate HIV-1 from latency. Sci. Rep. 2017, 7, 1–12.
- Zhang, X.X.; Lin, J.; Liang, T.Z.; Duan, H.; Tan, X.H.; Xi, B.M.; Li, L.; Liu, S.W. The BET bromodomain inhibitor apabetalone induces apoptosis of latent HIV-1 reservoir cells following viral reactivation. Acta Pharmacol. Sin. 2019, 40, 98–110.
- Liang, T.; Zhang, X.; Lai, F.; Lin, J.; Zhou, C.; Xu, X.; Tan, X.; Liu, S.; Li, L. A novel bromodomain inhibitor, CPI-203, serves as an HIV-1 latency-reversing agent by activating positive transcription elongation factor b. Biochem. Pharmacol. 2019, 164, 237–251.
- Zhao, M.; De Crignis, E.; Rokx, C.; Verbon, A.; van Gelder, T.; Mahmoudi, T.; Katsikis, P.D.; Mueller, Y.M. T cell toxicity of HIV latency reversing agents. Pharmacol. Res. 2019, 139, 524–534.
- Sánchez-Ventura, J.; Amo-Aparicio, J.; Navarro, X.; Penas, C. BET protein inhibition regulates cytokine production and promotes neuroprotection after spinal cord injury. J. Neuroinflammation 2019, 16, 1–12.
- Domínguez-Andrés, J.; Ferreira, A.V.; Jansen, T.; Smithers, N.; Prinjha, R.K.; Furze, R.C.; Netea, M.G. Bromodomain inhibitor I-BET151 suppresses immune responses during fungal–immune interaction. Eur. J. Immunol. 2019, 49, 2044–2050.
- Rueda, C.M.; Velilla, P.A.; Chougnet, C.A.; Montoya, C.J.; Rugeles, M.T. HIV-induced T-cell activation/exhaustion in rectal mucosa is controlled only partially by antiretroviral treatment. PLoS ONE 2012, 7, e30307.
- El-Far, M.; Halwani, R.; Said, E.; Trautmann, L.; Doroudchi, M.; Janbazian, L.; Fonseca, S.; van Grevenynghe, J.; Yassine-Diab, B.; Sékaly, R.P.; et al. T-cell exhaustion in HIV infection. Curr. HIV/AIDS Rep. 2008, 5, 13–19.
- Abner, E.; Stoszko, M.; Zeng, L.; Chen, H.C.; Izquierdo-Bouldstridge, A.; Konuma, T.; Zorita, E.; Fanunza, E.; Zhang, Q.; Mahmoudi, T.; et al. A new quinoline BRD4 inhibitor targets a distinct latent HIV-1 reservoir for reactivation from other “shock” drugs. J. Virol. 2018, 92, e02056-17.
- Huang, H.; Liu, S.; Jean, M.; Simpson, S.; Huang, H.; Merkley, M.; Hayashi, T.; Kong, W.; Rodríguez-Sánchez, I.; Zhang, X.; et al. A novel bromodomain inhibitor reverses HIV-1 latency through specific binding with BRD4 to promote Tat and P-TEFb association. Front. Microbiol. 2017, 8, 1035.
- Cheung, K.; Lu, G.; Sharma, R.; Vincek, A.; Zhang, R.; Plotnikov, A.N.; Zhang, F.; Zhang, Q.; Ju, Y.; Hu, Y.; et al. BET N-terminal bromodomain inhibition selectively blocks Th17 cell differentiation and ameliorates colitis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 2952–2957.
- Tanaka, M.; Roberts, J.M.; Seo, H.S.; Souza, A.; Paulk, J.; Scott, T.G.; DeAngelo, S.L.; Dhe-Paganon, S.; Bradner, J.E. Design and characterization of bivalent BET inhibitors. Nat. Chem. Biol. 2016, 12, 1089.
- Wang, Q.; Li, Y.; Xu, J.; Wang, Y.; Shi, D.; Liu, L.; Leung, E.L.H.; Yao, X. Computational study on the selective inhibition mechanism of MS402 to the first and second bromodomains of BRD4. Proteins 2019, 87, 3–11.

