Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Gur P. Kaushal | + 2465 word(s) | 2465 | 2021-06-03 10:12:15 | | | |
| 2 | Vivi Li | Meta information modification | 2465 | 2021-06-07 05:39:29 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kaushal, G.P. Autophagy in Kidney Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/10550 (accessed on 13 June 2026).
Kaushal GP. Autophagy in Kidney Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/10550. Accessed June 13, 2026.
Kaushal, Gur P.. "Autophagy in Kidney Disease" Encyclopedia, https://encyclopedia.pub/entry/10550 (accessed June 13, 2026).
Kaushal, G.P. (2021, June 05). Autophagy in Kidney Disease. In Encyclopedia. https://encyclopedia.pub/entry/10550
Kaushal, Gur P.. "Autophagy in Kidney Disease." Encyclopedia. Web. 05 June, 2021.
Copy Citation
Autophagy is a dynamic process by which intracellular damaged macromolecules and organelles are degraded and recycled for the synthesis of new cellular components. Basal autophagy in the kidney acts as a quality control system and is vital for cellular metabolic and organelle homeostasis. Under pathological conditions, autophagy facilitates cellular adaptation; however, activation of autophagy in response to renal injury may be insufficient to provide protection, especially under dysregulated conditions. Kidney-specific deletion of Atg genes in mice has consistently demonstrated worsened acute kidney injury (AKI) outcomes supporting the notion of a pro-survival role of autophagy.
autophagy
acute kidney injury
chronic kidney disease
diabetic nephropathy
mTORC1
AMPK
renal fibrosis
apoptosis
regulated necrosis
1. Introduction
Recent discoveries elucidating the molecular machinery of autophagy and its fundamental role in important cellular functions have demonstrated the relevance of autophagy in human diseases. The Nobel Committee for Physiology or Medicine recognized Yoshinori Ohsumi for his novel work in identifying the biological process of autophagy and defining its critical function [1], awarding him the Nobel prize in 2016. Therefore, autophagy research on the pathogenesis of numerous diseases, including renal injury, is an area of great interest that may provide valuable insights into potential therapeutic opportunities.
The process of autophagy involves the formation of a double-membrane structure known as an autophagosome, which first sequesters the cellular constituents and subsequently delivers them to the lysosome for degradation [2] (Figure 1). This dynamic process culminates in the recycling of degraded products for the biosynthesis of new cellular components and for meeting the energy needs of the cell [2]. Autophagy-lysosomal and ubiquitin-proteasomal systems are the two major evolutionarily conserved cellular degradation pathways that play pivotal roles in regulating cellular homeostasis and maintaining quality control mechanisms [3][4]. The ubiquitin-proteasomal pathway involves a multimeric proteasome for the degradation of ubiquitinated small and short-lived proteins, whereas the autophagy process uses lysosomal hydrolases to degrade large intracellular heterogeneous misfolded proteins, protein aggregates, and damaged macromolecules, and has the ability to degrade entire damaged organelles and invading microorganisms [3]. The details of the autophagy process are summarized in Figure 1.
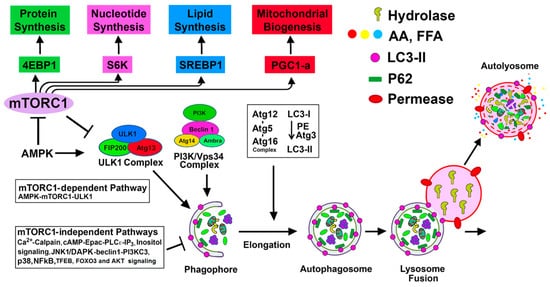
Figure 1. mTORC1-dependent and mTORC1-independent regulation of autophagy activation: Suppression of mTORC1 and induction of adenosine monophosphate activated protein kinase (AMPK) promote Unc-51 like autophagy activating kinase (ULK1) complex [ULK1, Atg13, focal adhesion kinase family interacting protein of 200 kD (FIP200), and Atg101] activation at the pre-autophagosomal assembly site (the certain domain of the ER) and initiates the autophagy process. Active ULK1 complex regulates the activity of the class III phosphatidylinositol (PtdIns) 3-kinase complex (including Beclin-1, Atg14(L)/barkor, Vps15, Vps34, and Ambra1) that generates phosphatidylinositol 3-phosphate (PI3P) rich domain. PI3P and PI3P-binding proteins (double FYVE domain-containing protein 1 (DFCP-1) and WIPI proteins) participate in the nucleation and associated membrane dynamics of the phagophore structure, the site of nucleation. Atg9 positive vesicles, that traffic from Golgi and endosomes and regulated by ULK1 complex, contribute to the formation of omegosomes and phagophores. Expansion and maturation of autophagosome from the phagophore structure require two ubiquitin-like conjugation systems that produce Atg12-Atg5-Atg16 oligomeric complex (Atg16 L1 complex) and lipidation of microtubule-associated protein 1A/1B-light chain 3 (LC3) (mammalian homolog of yeast Atg8) with phosphatidyl-ethanolamine (PE). WIPI upon binding to PI3P recruits Atg16 L1 complex to PI3P initiation sites. Atg12-Atg5 of the Atg L1 complex is then involved as an E3-like enzyme for the formation of the lipidated form of LC3. Once the autophagosome is produced, it then fuses with the lysosome to form autolysosome, and subsequently, the lysosomal hydrolases degrade the sequestered cargo. mTORC1 positively regulates protein synthesis, nucleotide synthesis, lipid synthesis, and mitochondrial biogenesis, as shown in the figure. mTORC1-independent alternative pathways negatively regulate autophagy.
2. Basal Autophagy in the Kidney and Other Organs
A basal level of autophagy fulfills a vital role in cellular metabolism and organelle homeostasis by degrading and recycling damaged proteins, macromolecules, and organelles. The importance of basal autophagy in cellular homeostasis in the kidney and other organs has been demonstrated in conditional autophagy-knockout (KO) mice (tissue-specific Atg5- or Atg7-KO mice) and autophagy-deficient cells. The accumulation of protein aggregates and inclusion bodies has been observed in autophagy-deficient renal tubular cells [5][6], hepatocytes [7][8], cardiomyocytes [9], and neural cells [10][11]. In addition, proximal tubule-specific autophagy-KO mice accumulated deformed or damaged mitochondria, p62, ubiquitin-positive inclusion bodies, and misfolded protein aggregates; and they exhibited increased proximal tubule cell apoptosis [5]. These mice at 24 months of age exhibited multiple characteristics of cellular senescence, including renal function loss, mitochondrial damage, nuclear DNA damage, and fibrosis [6], compared to wild-type mice. Moreover, podocyte-specific Atg5-deficient mice were found to develop mild albuminuria, podocyte loss, late-onset glomerulosclerosis, accumulation of oxidized and ubiquitinated protein aggregates, endoplasmic reticulum (ER) stress, and proteinuria [12]. Altogether, these findings indicate the pivotal role of basal autophagy in cellular remodeling and intracellular homeostasis in both tubules and podocytes.
3. Autophagy in Response to Stress and Kidney Injury
Autophagy activation is an adaptive response to a wide variety of internal and external cellular stresses (e.g., cell starvation, hypoxia, nutrient and growth factor deprivation, oxidant injury, genotoxic agents, and other damaging insults) as well as pathological conditions. Stress-induced autophagy can support cell survival by eliminating and recycling damaged macromolecules, protein aggregates, and organelles [13][14][15][16][17]. Furthermore, in response to stress, organelle-specific autophagy, known as selective autophagy, eliminates and recycles damaged organelles, including mitochondria, peroxisomes, lysosomes, ER, and even the nucleus [18]. Although stress-induced autophagy is considered to be primarily protective [15], its ultimate effect on cell survival may depend on the relative activation and completion of the autophagy process. Ideally, optimal autophagic flux ensures an autophagy-mediated supply of bioenergy molecules (e.g., ATP) and break down products (e.g., amino acids and fatty acids) to maintain cellular biosynthesis. The pro-survival function may depend on the autophagic suppression of the stress-mediated cell death pathways, including apoptosis and regulated necrosis [19][20][21].
Indeed, dysregulation or failure of the autophagy pathway or mutations in the autophagy-related genes (Atg genes) results in various human pathologies, including cancer, neurodegenerative diseases, chronic inflammatory diseases, and cardiac failure [22][23][24]. Autophagy may also promote cell death under some special circumstances. It has been suggested that high levels of autophagy may cause excessive digestion of cellular constituents, resulting in cell death. For example, a high level of autophagy induction by the cell-permeable peptide transactivator of transcription (TAT)-beclin-1 derived from beclin-1 in cell cultures causes cell death [25]. Autophagic cell death, called ‘autosis’, is a nonapoptotic cell death mechanism triggered by hypoxia, starvation, or cell-permeable beclin-1-derived autophagy-inducing peptides and is regulated by the Na-K-ATPase pump [25]. Moreover, cell death by autophagy is promoted by reactive oxygen species produced upon degradation of ferritin by autophagy, a process known as ferroptosis [26].
Renal tubular epithelial cells under injury conditions are exposed to multiple stresses, including oxidative stress, hypoxia, nutrient and energy depletion, endoplasmic reticulum (ER) stress, mitochondrial damage, and genotoxic stress, all of which can activate autophagy. However, insufficient or defective autophagy due to impaired clearance of damaged macromolecules and organelles is unable to provide protection from cellular stress in acute kidney injury (AKI) and other renal diseases. The specific role of autophagy in models of AKI and progressive renal disease has been revealed by using both pharmacological and genetic approaches (described below).
3.1. Autophagy in AKI
Autophagy is activated in the kidney in AKI induced by ischemia-reperfusion (IR), cisplatin, and sepsis. The role of autophagy in AKI using both genetic and pharmacological approaches has been recently reviewed [27][28][29]. Conditional proximal tubule-specific Atg5- or Atg7-KO mice subjected to IR, cisplatin, or sepsis-induced AKI have consistently demonstrated worsened outcomes compared to wild-type mice, supporting a pro-survival role of autophagy in models of AKI [27][28]. Compared to control mice, proximal tubule-specific autophagy-deficient mice exhibited increased tubular damage, loss of renal function, tubular cell apoptosis, mitochondrial damage, and accumulation of p62 and ubiquitin-positive inclusion bodies in response to IR [5][6][30][31]. However, mice with conditional selective deletion of Atg5 from the proximal tubular S3 segment exhibited a sharp rise in cell death (TUNEL positive cells but no increase in caspase-3 activation) at 2 h after IR, but less tubular damage and inflammation 3 days later compared to normal mice [32]. Hence, the outcome of IR injury differs depending on whether Atg5 is deleted from the S3 segment alone versus from all three segments (S1, S2, and S3) of the tubule [5][30]. An increase in the TUNEL positive tubular cells with an increase of caspase-3 activity in mice deficient in Atg5 in all segments, as well as in mice with Atg5 deleted in the S3 segment alone without caspase-3 activation, suggests the involvement of different pathways of cell death [33]. Different modes of cell death, including apoptosis and regulated necrosis (necroptosis, ferroptosis, and parthanatos as described below), have been recently reported to occur during AKI [27][34]. Since autophagy inhibition by pharmacological approaches activates cell death pathways in renal [27][35], as well as in non-renal cells [19][36], the pro-survival effect of autophagy activation should then influence the interplay between autophagy and different cell death pathways and influence the cell fate.
3.2. Autophagy in Renal Interstitial Fibrosis and Progressive Kidney Disease
A hallmark of chronic kidney disease (CKD) is a progressive deposition of extracellular matrix proteins, which correlate well with the deterioration of renal function, regardless of the etiology of the primary insult [37][38][39]. In addition to various causes of CKD, acute kidney injury (AKI) is also a major contributing factor in the progression of CKD due to abnormal post-AKI recovery and ensuing progressive fibrosis, leading to end-stage renal disease (ESRD) [40][41].
To determine the role of autophagy in renal fibrosis, most studies have used the unilateral ureteral obstruction (UUO) model [42]; this model exhibits time-dependent induction of autophagy accompanied by tubular atrophy, tubular cell death, and interstitial fibrosis [43][44]. The autophagy inhibitor 3-methyladenine (3-MA) enhanced tubular apoptosis and interstitial fibrosis in obstructed kidneys [44]. In addition, transgenic mice with heterozygous deletion of beclin-1 (beclin-1 ± mice) showed increased deposition of type-1 collagen [45]. LC3)-KO and beclin-1 ± mice subjected to the UUO model revealed increased deposition of collagen accompanied by increased levels of transforming growth factor (TGF)-β1 in the obstructed kidney [46]. In this model, the induction of autophagy in distal tubular epithelial cells afforded protection from renal tubulointerstitial fibrosis through the regulation of the TGF-β/signal transducers and transcriptional modulator (Smad)4 signaling pathway and NOD-LRR and pyrin domain-containing protein3 (NLRP3) inflammasome/caspase-1/interleukin-1 β (IL-1β) signaling pathway [47]. Conditional deletion of Atg5 enhanced renal interstitial fibrosis and promoted cell cycle arrest at G2/M [48]. It was shown that the deletion of ATG5 in proximal tubular epithelial cells promoted leukocyte infiltration and expression of proinflammatory cytokines, while overexpression of ATG5 inhibited the inflammatory response in an autophagy-dependent manner via blocking nuclear factor kappa-light chain enhancer of activated B cells (NF-κB) signaling that provides protection against renal inflammation and accompanied fibrosis [49].
A recent study showed that phosphatase and tension homologue (PTEN)-induced kinase 1/ (PINK1)/mitofusion 2 (MFN2)/Parkin-mediated macrophage mitophagy is downregulated during kidney fibrosis, and loss of either Pink1 or Parkin promoted macrophage development toward profibrotic/M2 macrophages and subsequent renal fibrosis [50]. Additionally, autophagy induced by the histone deacetylase inhibitor, valproic acid, suppressed renal fibrosis in mice subjected to UUO [51]. Taken together, these studies support that autophagy suppresses renal fibrosis in obstructed kidneys and may provide a pro-survival role (Table 1). Rubicon, a negative regulator of autophagy, increased during aging, suppressed autophagic activity, and caused fibrosis in mouse kidney and α-Syn accumulation in mouse brain [52]. In streptozotocin (STZ)-induced diabetic nephropathy in rats, microRNA (miR)-22 upregulation was associated with increased fibrosis and suppression of autophagy [53]. In normal rat kidney (NRK)-52E cells, rapamycin-induced autophagy reduced high glucose-induced collagen IV (Col IV), and α–smooth muscle actin (α-SMA) expression and overexpression of miR-22 suppressed autophagic flux and induced the expression of Col IV and α-SMA [53]. Also, triptolide (TP), a traditional Chinese medicine, reduced fibrosis by increasing autophagy via the miR-141-3p/PTEN/protein kinase B (Akt)/mTOR pathway in STZ-induced diabetic nephropathy in high fat diet (HFD)-fed rats [54]. 1,25-dihydroxyvitamin D3 ameliorated Ang II-induced tubulointerstitial fibrosis, expanded mesangial regions and foot process fusion, and impaired autophagy by improving mitochondrial dysfunction and by modulating autophagy [55]. Elafibranor, a novel dual peroxisome proliferator-activator receptor α/δ (PPARα/δ) agonist, protected HFD mice with CKD by improving kidney-specific protective effects, including preservation of glomerular/tubular barrier protein, maintenance of the structure, antioxidative stress, and activation of sirtuin (SIRT)-autophagy [56]. Postconditioning (POC) following IR injury reduced renal damage and renal fibrosis by increased autophagy [57]. Periostin gene, also known as osteoblast-specific factor-2 that plays a role as a profibrotic and proinflammatory factor [58], is upregulated in kidneys with 5-6 nephrectomy and impaired autophagy flux. Knockdown of periostin afforded protection against 5/6 nephrectomy-induced intrarenal renin-angiotensin system activation, fibrosis, inflammation in rats, and improved autophagy flux [59], suggesting that periostin-induced impaired autophagy is involved in the inflammation and fibrosis in the profibrotic model. In contrast, other studies have recently shown that the induction of autophagy results in renal fibrosis. Persistent activation of autophagy during UUO promoted renal interstitial fibrosis, macrophage infiltration, and tubular atrophy [60]. Proximal tubule-specific deletion of Atg7 suppressed tubular atrophy, nephron loss, interstitial macrophage infiltration, interstitial fibrosis, and expression of the profibrotic factor fibroblast growth factor 2 (FGF2) [60]. Furthermore, a Chinese herb rhubarb and its bioactive component rhein that improved renal function and the glomerular filtration rate (GFR) in stage 3 and 4 patients with CKD [61] inhibited autophagy. Protein kinase Cα (PKCα) is activated during UUO fibrotic kidney, and inhibition of PKCα blocked autophagic flux in fibroblasts of the fibrotic kidneys and prevented fibroblast activation and kidney fibrosis [62]. Rhubarb also suppressed renal fibrosis [63]. These studies are summarized in Table 1. In view of the above results, additional studies with different experimental models of renal fibrosis are required using both genetic and pharmacological approaches to better understand the definitive role of autophagy in renal interstitial fibrosis.
Table 1. Autophagy in renal interstitial fibrosis.
| Kidney Disease | Agent/Drug | Effect on Autophagy | Effect on Fibrosis | Reference |
|---|---|---|---|---|
| Autophagy suppresses fibrosis | ||||
| UUO model | 3-MA | ↓ autophagy | ↑ in interstitial fibrosis and tubular apoptosis | [42][43][44] |
| LC3 KO and beclin-1 ± |
↓ autophagy | ↑ deposition of collagen and TGF-β1 | [46] | |
| Conditional deletion of ATG7 in distal tubule | ↓ autophagy in the distal tubules | ↑ in tubulointerstitial fibrosis via the TGF-β/Smad4 and NLRP3 signaling | [47] | |
| Conditional deletion of ATG5 | ↓ autophagy | ↑ renal interstitial fibrosis and cell cycle arrest at G2/M | [48] | |
| Proximal tubule specific deletion of ATG5 | ↓ autophagy | ↑ renal fibrosis due to leukocyte infiltration and expression of pro-inflammatory cytokines | [49] | |
| Valproic acid (histone deacetylase inhibitor) | ↑ autophagy | ↓ in renal fibrosis | [51] | |
| Rubicon | ↓ autophagy | ↑ in renal fibrosis | [52] | |
| STZ- DN | miR -22 upregulation | ↓ autophagy | ↑ in renal fibrosis with increased expression of col-IV and α-SMA | [53] |
| Triptolide | ↑ autophagy via miR-141-3p/PTEN/Akt/mTOR pathway | ↓ in renal fibrosis | [54] | |
| HFD with CKD | Elafibranor (dual PPARα/δ agonist) | ↑ autophagy mediated by SIRT1 | ↓ in renal fibrosis | [56] |
| 5/6-Nephrectomy | Knockdown of periostin gene (osteoblast specific factor-2) | ↑ autophagy and upregulates periostin gene (pro-fibrotic and pro-inflammatory factor) | ↓ in renal inflammation and fibrosis | [58] |
| Proximal tubule specific deletion of ATG7 | ↑ autophagy | ↓ in renal fibrosis with pro-fibrotic FGF2 | [60] | |
| Autophagy promotes fibrosis | ||||
| Ang II- induced CKD | 1,25-dihydroxyvitamin D3 | ↓ autophagy with improved mitochondrial dysfunction | ↓ in renal fibrosis | [55] |
| Stage 3 and stage 4 CKD | Rhubarb (Rhein- bioactive component) | ↓ autophagy | ↓ in renal fibrosis | [61] |
References
- Levine, B.; Klionsky, D.J. Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: Breakthroughs in baker’s yeast fuel advances in biomedical research. Proc. Natl. Acad. Sci. USA 2017, 114, 201–205.
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588–596.
- Korolchuk, V.I.; Menzies, F.M.; Rubinsztein, D.C. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010, 584, 1393–1398.
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224.
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Kaimori, J.Y.; Matsui, I.; Namba, T.; Kitamura, H.; Niimura, F.; Matsusaka, T.; Soga, T.; et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J. Am. Soc. Nephrol. 2011, 22, 902–913.
- Yamamoto, T.; Takabatake, Y.; Kimura, T.; Takahashi, A.; Namba, T.; Matsuda, J.; Minami, S.; Kaimori, J.Y.; Matsui, I.; Kitamura, H.; et al. Time-dependent dysregulation of autophagy: Implications in aging and mitochondrial homeostasis in the kidney proximal tubule. Autophagy 2016, 12, 801–813.
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434.
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800.
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007, 13, 619–624.
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889.
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884.
- Hartleben, B.; Godel, M.; Meyer-Schwesinger, C.; Liu, S.; Ulrich, T.; Kobler, S.; Wiech, T.; Grahammer, F.; Arnold, S.J.; Lindenmeyer, M.T.; et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J. Clin. Investig. 2010, 120, 1084–1096.
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293.
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741.
- Murrow, L.; Debnath, J. Autophagy as a stress-response and quality-control mechanism: Implications for cell injury and human disease. Annu. Rev. Pathol. 2013, 8, 105–137.
- Galluzzi, L.; Bravo-San Pedro, J.M.; Kepp, O.; Kroemer, G. Regulated cell death and adaptive stress responses. Cell Mol. Life Sci. 2016, 73, 2405–2410.
- Ktistakis, N.T.; Tooze, S.A. Digesting the Expanding Mechanisms of Autophagy. Trends Cell Biol. 2016, 26, 624–635.
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-Dependent and Independent Signals In Selective Autophagy. Trends Cell Biol. 2016, 26, 6–16.
- Anding, A.L.; Baehrecke, E.H. Autophagy in Cell Life and Cell Death. Curr. Top. Dev. Biol. 2015, 114, 67–91.
- Fitzwalter, B.E.; Thorburn, A. Recent insights into cell death and autophagy. FEBS J. 2015, 282, 4279–4288.
- Altman, B.J.; Rathmell, J.C. Metabolic stress in autophagy and cell death pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a008763.
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 1845–1846.
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell Res 2014, 24, 69–79.
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511.
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M., Jr.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371.
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., III; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428.
- Kaushal, G.P.; Shah, S.V. Autophagy in acute kidney injury. Kidney Int. 2016, 89, 779–791.
- Livingston, M.J.; Dong, Z. Autophagy in acute kidney injury. Semin. Nephrol. 2014, 34, 17–26.
- Choi, M.E. Autophagy in Kidney Disease. Annu. Rev. Physiol. 2019.
- Liu, S.; Hartleben, B.; Kretz, O.; Wiech, T.; Igarashi, P.; Mizushima, N.; Walz, G.; Huber, T.B. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 2012, 8, 826–837.
- Jiang, M.; Wei, Q.; Dong, G.; Komatsu, M.; Su, Y.; Dong, Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012, 82, 1271–1283.
- Baisantry, A.; Bhayana, S.; Rong, S.; Ermeling, E.; Wrede, C.; Hegermann, J.; Pennekamp, P.; Sorensen-Zender, I.; Haller, H.; Melk, A.; et al. Autophagy Induces Prosenescent Changes in Proximal Tubular S3 Segments. J. Am. Soc. Nephrol. 2016, 27, 1609–1616.
- Kers, J.; Leemans, J.C.; Linkermann, A. An Overview of Pathways of Regulated Necrosis in Acute Kidney Injury. Semin. Nephrol. 2016, 36, 139–152.
- Linkermann, A.; Chen, G.; Dong, G.; Kunzendorf, U.; Krautwald, S.; Dong, Z. Regulated cell death in AKI. J. Am. Soc. Nephrol. 2014, 25, 2689–2701.
- Havasi, A.; Dong, Z. Autophagy and Tubular Cell Death in the Kidney. Semin. Nephrol. 2016, 36, 174–188.
- Boya, P.; Gonzalez-Polo, R.A.; Casares, N.; Perfettini, J.L.; Dessen, P.; Larochette, N.; Metivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell Biol. 2005, 25, 1025–1040.
- Bohle, A.; Strutz, F.; Muller, G.A. On the pathogenesis of chronic renal failure in primary glomerulopathies: A view from the interstitium. Exp. Nephrol. 1994, 2, 205–210.
- Nath, K.A. Tubulointerstitial changes as a major determinant in the progression of renal damage. Am. J. Kidney Dis. 1992, 20, 1–17.
- Boor, P.; Ostendorf, T.; Floege, J. Renal fibrosis: Novel insights into mechanisms and therapeutic targets. Nat. Rev. Nephrol. 2010, 6, 643–656.
- Belayev, L.Y.; Palevsky, P.M. The link between acute kidney injury and chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2014, 23, 149–154.
- Venkatachalam, M.A.; Weinberg, J.M.; Kriz, W.; Bidani, A.K. Failed Tubule Recovery, AKI-CKD Transition, and Kidney Disease Progression. J. Am. Soc. Nephrol. 2015, 26, 1765–1776.
- Chevalier, R.L.; Forbes, M.S.; Thornhill, B.A. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 2009, 75, 1145–1152.
- Li, L.; Zepeda-Orozco, D.; Black, R.; Lin, F. Autophagy is a component of epithelial cell fate in obstructive uropathy. Am. J. Pathol. 2010, 176, 1767–1778.
- Kim, W.Y.; Nam, S.A.; Song, H.C.; Ko, J.S.; Park, S.H.; Kim, H.L.; Choi, E.J.; Kim, Y.S.; Kim, J.; Kim, Y.K. The role of autophagy in unilateral ureteral obstruction rat model. Nephrology 2012, 17, 148–159.
- Kim, S.I.; Na, H.J.; Ding, Y.; Wang, Z.; Lee, S.J.; Choi, M.E. Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-β1. J. Biol. Chem. 2012, 287, 11677–11688.
- Ding, Y.; Kim, S.; Lee, S.Y.; Koo, J.K.; Wang, Z.; Choi, M.E. Autophagy regulates TGF-β expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J. Am. Soc. Nephrol. 2014, 25, 2835–2846.
- Nam, S.A.; Kim, W.-Y.; Kim, J.W.; Park, S.H.; Kim, H.L.; Lee, M.-S.; Komatsu, M.; Ha, H.; Lim, J.H.; Park, C.W.; et al. Autophagy attenuates tubulointerstital fibrosis through regulating transforming growth factor-β and NLRP3 inflammasome signaling pathway. Cell Death Dis. 2019, 10, 78.
- Li, H.; Peng, X.; Wang, Y.; Cao, S.; Xiong, L.; Fan, J.; Wang, Y.; Zhuang, S.; Yu, X.; Mao, H. Atg5-mediated autophagy deficiency in proximal tubules promotes cell cycle G2/M arrest and renal fibrosis. Autophagy 2016, 12, 1472–1486.
- Peng, X.; Wang, Y.; Li, H.; Fan, J.; Shen, J.; Yu, X.; Zhou, Y.; Mao, H. ATG5-mediated autophagy suppresses NF-κB signaling to limit epithelial inflammatory response to kidney injury. Cell Death Dis. 2019, 10, 253.
- Bhatia, D.; Chung, K.P.; Nakahira, K.; Patino, E.; Rice, M.C.; Torres, L.K.; Muthukumar, T.; Choi, A.M.; Akchurin, O.M.; Choi, M.E. Mitophagy dependent macrophage reprogramming protects against kidney fibrosis. JCI Insight 2019.
- Kawaoka, K.; Doi, S.; Nakashima, A.; Yamada, K.; Ueno, T.; Doi, T.; Masaki, T. Valproic acid attenuates renal fibrosis through the induction of autophagy. Clin. Exp. Nephrol. 2017, 21, 771–780.
- Nakamura, S.; Oba, M.; Suzuki, M.; Takahashi, A.; Yamamuro, T.; Fujiwara, M.; Ikenaka, K.; Minami, S.; Tabata, N.; Yamamoto, K.; et al. Suppression of autophagic activity by Rubicon is a signature of aging. Nat. Commun. 2019, 10, 847.
- Zhang, Y.; Zhao, S.; Wu, D.; Liu, X.; Shi, M.; Wang, Y.; Zhang, F.; Ding, J.; Xiao, Y.; Guo, B. MicroRNA-22 Promotes Renal Tubulointerstitial Fibrosis by Targeting PTEN and Suppressing Autophagy in Diabetic Nephropathy. J. Diabetes Res. 2018, 2018, 4728645.
- Li, X.-Y.; Wang, S.-S.; Han, Z.; Han, F.; Chang, Y.-P.; Yang, Y.; Xue, M.; Sun, B.; Chen, L.-M. Triptolide Restores Autophagy to Alleviate Diabetic Renal Fibrosis through the miR-141-3p/PTEN/Akt/mTOR Pathway. Mol. Ther. Nucleic Acids 2017, 9, 48–56.
- Shen, Q.; Bi, X.; Ling, L.; Ding, W. 1,25-Dihydroxyvitamin D3 Attenuates Angiotensin II-Induced Renal Injury by Inhibiting Mitochondrial Dysfunction and Autophagy. Cell. Physiol. Biochem. 2018, 51, 1751–1762.
- Tsai, H.C.; Chang, F.P.; Li, T.H.; Liu, C.W.; Huang, C.C.; Huang, S.F.; Yang, Y.Y.; Lee, K.C.; Hsieh, Y.C.; Wang, Y.W.; et al. Elafibranor Inhibits Chronic Kidney Disease Progression in NASH Mice. BioMed Res. Int. 2019, 2019, 6740616.
- Song, Y.; Tao, Q.; Yu, L.; Li, L.; Bai, T.; Song, X.; Hu, H.; Li, Y.; Tan, X. Activation of autophagy contributes to the renoprotective effect of postconditioning on acute kidney injury and renal fibrosis. Biochem. Biophys. Res. Commun. 2018, 504, 641–646.
- Kudo, A.; Kii, I. Periostin function in communication with extracellular matrices. J. Cell Commun. Signal. 2018, 12, 301–308.
- Bian, X.; Bai, Y.; Su, X.; Zhao, G.; Sun, G.; Li, D. Knockdown of periostin attenuates 5/6 nephrectomy-induced intrarenal renin-angiotensin system activation, fibrosis, and inflammation in rats. J. Cell Physiol. 2019, 234, 22857–22873.
- Livingston, M.J.; Ding, H.F.; Huang, S.; Hill, J.A.; Yin, X.M.; Dong, Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy 2016, 12, 976–998.
- Khan, I.A.; Nasiruddin, M.; Haque, S.F.; Khan, R.A. Evaluation of Rhubarb Supplementation in Stages 3 and 4 of Chronic Kidney Disease: A Randomized Clinical Trial. Int. J. Chronic Dis. 2014, 2014, 789340.
- Xue, X.; Ren, J.; Sun, X.; Gui, Y.; Feng, Y.; Shu, B.; Wei, W.; Lu, Q.; Liang, Y.; He, W.; et al. Protein kinase Cα drives fibroblast activation and kidney fibrosis by stimulating autophagic flux. J. Biol. Chem. 2018, 293, 11119–11130.
- Tu, Y.; Gu, L.; Chen, D.; Wu, W.; Liu, H.; Hu, H.; Wan, Y.; Sun, W. Rhein Inhibits Autophagy in Rat Renal Tubular Cells by Regulation of AMPK/mTOR Signaling. Sci. Rep. 2017, 7, 43790.
More
Information
Subjects:
Cell Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Revisions:
2 times
(View History)
Update Date:
07 Jun 2021
Table of Contents




Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No