Autophagy is a dynamic process by which intracellular damaged macromolecules and organelles are degraded and recycled for the synthesis of new cellular components. Basal autophagy in the kidney acts as a quality control system and is vital for cellular metabolic and organelle homeostasis. Under pathological conditions, autophagy facilitates cellular adaptation; however, activation of autophagy in response to renal injury may be insufficient to provide protection, especially under dysregulated conditions. Kidney-specific deletion of Atg genes in mice has consistently demonstrated worsened acute kidney injury (AKI) outcomes supporting the notion of a pro-survival role of autophagy.
- autophagy
- acute kidney injury
- chronic kidney disease
- diabetic nephropathy
- mTORC1
- AMPK
- renal fibrosis
- apoptosis
- regulated necrosis
1. Introduction
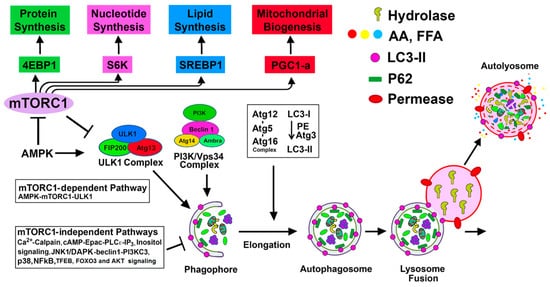
2. Basal Autophagy in the Kidney and Other Organs
3. Autophagy in Response to Stress and Kidney Injury
3.1. Autophagy in AKI
3.2. Autophagy in Renal Interstitial Fibrosis and Progressive Kidney Disease
| Kidney Disease | Agent/Drug | Effect on Autophagy | Effect on Fibrosis | Reference |
|---|---|---|---|---|
| Autophagy suppresses fibrosis | ||||
| UUO model | 3-MA | ↓ autophagy | ↑ in interstitial fibrosis and tubular apoptosis | [42,43,44] |
| LC3 KO and beclin-1 ± |
↓ autophagy | ↑ deposition of collagen and TGF-β1 | [46] | |
| Conditional deletion of ATG7 in distal tubule | ↓ autophagy in the distal tubules | ↑ in tubulointerstitial fibrosis via the TGF-β/Smad4 and NLRP3 signaling | [47] | |
| Conditional deletion of ATG5 | ↓ autophagy | ↑ renal interstitial fibrosis and cell cycle arrest at G2/M | [48] | |
| Proximal tubule specific deletion of ATG5 | ↓ autophagy | ↑ renal fibrosis due to leukocyte infiltration and expression of pro-inflammatory cytokines | [49] | |
| Valproic acid (histone deacetylase inhibitor) | ↑ autophagy | ↓ in renal fibrosis | [51] | |
| Rubicon | ↓ autophagy | ↑ in renal fibrosis | [52] | |
| STZ- DN | miR -22 upregulation | ↓ autophagy | ↑ in renal fibrosis with increased expression of col-IV and α-SMA | [53] |
| Triptolide | ↑ autophagy via miR-141-3p/PTEN/Akt/mTOR pathway | ↓ in renal fibrosis | [54] | |
| HFD with CKD | Elafibranor (dual PPARα/δ agonist) | ↑ autophagy mediated by SIRT1 | ↓ in renal fibrosis | [56] |
| 5/6-Nephrectomy | Knockdown of periostin gene (osteoblast specific factor-2) | ↑ autophagy and upregulates periostin gene (pro-fibrotic and pro-inflammatory factor) | ↓ in renal inflammation and fibrosis | [58] |
| Proximal tubule specific deletion of ATG7 | ↑ autophagy | ↓ in renal fibrosis with pro-fibrotic FGF2 | [60] | |
| Autophagy promotes fibrosis | ||||
| Ang II- induced CKD | 1,25-dihydroxyvitamin D3 | ↓ autophagy with improved mitochondrial dysfunction | ↓ in renal fibrosis | [55] |
| Stage 3 and stage 4 CKD | Rhubarb (Rhein- bioactive component) | ↓ autophagy | ↓ in renal fibrosis | [61] |
This entry is adapted from the peer-reviewed paper 10.3390/biom10010100