Autophagy is a dynamic process by which intracellular damaged macromolecules and organelles are degraded and recycled for the synthesis of new cellular components. Basal autophagy in the kidney acts as a quality control system and is vital for cellular metabolic and organelle homeostasis. Under pathological conditions, autophagy facilitates cellular adaptation; however, activation of autophagy in response to renal injury may be insufficient to provide protection, especially under dysregulated conditions. Kidney-specific deletion of Atg genes in mice has consistently demonstrated worsened acute kidney injury (AKI) outcomes supporting the notion of a pro-survival role of autophagy.
- autophagy
- acute kidney injury
- chronic kidney disease
- diabetic nephropathy
- mTORC1
- AMPK
- renal fibrosis
- apoptosis
- regulated necrosis
1. Introduction
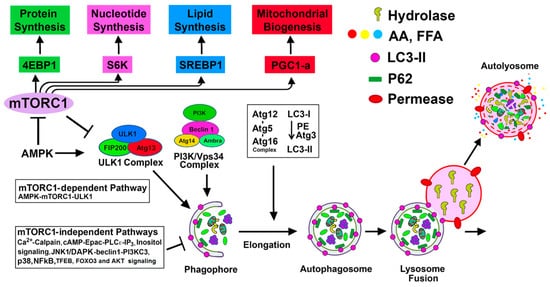
2. Basal Autophagy in the Kidney and Other Organs
3. Autophagy in Response to Stress and Kidney Injury
3.1. Autophagy in AKI
3.2. Autophagy in Renal Interstitial Fibrosis and Progressive Kidney Disease
| Kidney Disease | Agent/Drug | Effect on Autophagy | Effect on Fibrosis | Reference |
|---|---|---|---|---|
| Autophagy suppresses fibrosis | ||||
| UUO model | 3-MA | ↓ autophagy | ↑ in interstitial fibrosis and tubular apoptosis | [42][43][44] |
| LC3 KO and beclin-1 ± |
↓ autophagy | ↑ deposition of collagen and TGF-β1 | [46] | |
| Conditional deletion of ATG7 in distal tubule | ↓ autophagy in the distal tubules | ↑ in tubulointerstitial fibrosis via the TGF-β/Smad4 and NLRP3 signaling | [47] | |
| Conditional deletion of ATG5 | ↓ autophagy | ↑ renal interstitial fibrosis and cell cycle arrest at G2/M | [48] | |
| Proximal tubule specific deletion of ATG5 | ↓ autophagy | ↑ renal fibrosis due to leukocyte infiltration and expression of pro-inflammatory cytokines | [49] | |
| Valproic acid (histone deacetylase inhibitor) | ↑ autophagy | ↓ in renal fibrosis | [51] | |
| Rubicon | ↓ autophagy | ↑ in renal fibrosis | [52] | |
| STZ- DN | miR -22 upregulation | ↓ autophagy | ↑ in renal fibrosis with increased expression of col-IV and α-SMA | [53] |
| Triptolide | ↑ autophagy via miR-141-3p/PTEN/Akt/mTOR pathway | ↓ in renal fibrosis | [54] | |
| HFD with CKD | Elafibranor (dual PPARα/δ agonist) | ↑ autophagy mediated by SIRT1 | ↓ in renal fibrosis | [56] |
| 5/6-Nephrectomy | Knockdown of periostin gene (osteoblast specific factor-2) | ↑ autophagy and upregulates periostin gene (pro-fibrotic and pro-inflammatory factor) | ↓ in renal inflammation and fibrosis | [58] |
| Proximal tubule specific deletion of ATG7 | ↑ autophagy | ↓ in renal fibrosis with pro-fibrotic FGF2 | [60] | |
| Autophagy promotes fibrosis | ||||
| Ang II- induced CKD | 1,25-dihydroxyvitamin D3 | ↓ autophagy with improved mitochondrial dysfunction | ↓ in renal fibrosis | [55] |
| Stage 3 and stage 4 CKD | Rhubarb (Rhein- bioactive component) | ↓ autophagy | ↓ in renal fibrosis | [61] |
This entry is adapted from the peer-reviewed paper 10.3390/biom10010100
References
- Levine, B.; Klionsky, D.J. Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: Breakthroughs in baker’s yeast fuel advances in biomedical research. Proc. Natl. Acad. Sci. USA 2017, 114, 201–205.
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588–596.
- Korolchuk, V.I.; Menzies, F.M.; Rubinsztein, D.C. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010, 584, 1393–1398.
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224.
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Kaimori, J.Y.; Matsui, I.; Namba, T.; Kitamura, H.; Niimura, F.; Matsusaka, T.; Soga, T.; et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J. Am. Soc. Nephrol. 2011, 22, 902–913.
- Yamamoto, T.; Takabatake, Y.; Kimura, T.; Takahashi, A.; Namba, T.; Matsuda, J.; Minami, S.; Kaimori, J.Y.; Matsui, I.; Kitamura, H.; et al. Time-dependent dysregulation of autophagy: Implications in aging and mitochondrial homeostasis in the kidney proximal tubule. Autophagy 2016, 12, 801–813.
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434.
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800.
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007, 13, 619–624.
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889.
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884.
- Hartleben, B.; Godel, M.; Meyer-Schwesinger, C.; Liu, S.; Ulrich, T.; Kobler, S.; Wiech, T.; Grahammer, F.; Arnold, S.J.; Lindenmeyer, M.T.; et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J. Clin. Investig. 2010, 120, 1084–1096.
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293.
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741.
- Murrow, L.; Debnath, J. Autophagy as a stress-response and quality-control mechanism: Implications for cell injury and human disease. Annu. Rev. Pathol. 2013, 8, 105–137.
- Galluzzi, L.; Bravo-San Pedro, J.M.; Kepp, O.; Kroemer, G. Regulated cell death and adaptive stress responses. Cell Mol. Life Sci. 2016, 73, 2405–2410.
- Ktistakis, N.T.; Tooze, S.A. Digesting the Expanding Mechanisms of Autophagy. Trends Cell Biol. 2016, 26, 624–635.
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-Dependent and Independent Signals In Selective Autophagy. Trends Cell Biol. 2016, 26, 6–16.
- Anding, A.L.; Baehrecke, E.H. Autophagy in Cell Life and Cell Death. Curr. Top. Dev. Biol. 2015, 114, 67–91.
- Fitzwalter, B.E.; Thorburn, A. Recent insights into cell death and autophagy. FEBS J. 2015, 282, 4279–4288.
- Altman, B.J.; Rathmell, J.C. Metabolic stress in autophagy and cell death pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a008763.
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 1845–1846.
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell Res 2014, 24, 69–79.
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511.
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M., Jr.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371.
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., III; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428.
- Kaushal, G.P.; Shah, S.V. Autophagy in acute kidney injury. Kidney Int. 2016, 89, 779–791.
- Livingston, M.J.; Dong, Z. Autophagy in acute kidney injury. Semin. Nephrol. 2014, 34, 17–26.
- Choi, M.E. Autophagy in Kidney Disease. Annu. Rev. Physiol. 2019.
- Liu, S.; Hartleben, B.; Kretz, O.; Wiech, T.; Igarashi, P.; Mizushima, N.; Walz, G.; Huber, T.B. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 2012, 8, 826–837.
- Jiang, M.; Wei, Q.; Dong, G.; Komatsu, M.; Su, Y.; Dong, Z. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 2012, 82, 1271–1283.
- Baisantry, A.; Bhayana, S.; Rong, S.; Ermeling, E.; Wrede, C.; Hegermann, J.; Pennekamp, P.; Sorensen-Zender, I.; Haller, H.; Melk, A.; et al. Autophagy Induces Prosenescent Changes in Proximal Tubular S3 Segments. J. Am. Soc. Nephrol. 2016, 27, 1609–1616.
- Kers, J.; Leemans, J.C.; Linkermann, A. An Overview of Pathways of Regulated Necrosis in Acute Kidney Injury. Semin. Nephrol. 2016, 36, 139–152.
- Linkermann, A.; Chen, G.; Dong, G.; Kunzendorf, U.; Krautwald, S.; Dong, Z. Regulated cell death in AKI. J. Am. Soc. Nephrol. 2014, 25, 2689–2701.
- Havasi, A.; Dong, Z. Autophagy and Tubular Cell Death in the Kidney. Semin. Nephrol. 2016, 36, 174–188.
- Boya, P.; Gonzalez-Polo, R.A.; Casares, N.; Perfettini, J.L.; Dessen, P.; Larochette, N.; Metivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell Biol. 2005, 25, 1025–1040.
- Bohle, A.; Strutz, F.; Muller, G.A. On the pathogenesis of chronic renal failure in primary glomerulopathies: A view from the interstitium. Exp. Nephrol. 1994, 2, 205–210.
- Nath, K.A. Tubulointerstitial changes as a major determinant in the progression of renal damage. Am. J. Kidney Dis. 1992, 20, 1–17.
- Boor, P.; Ostendorf, T.; Floege, J. Renal fibrosis: Novel insights into mechanisms and therapeutic targets. Nat. Rev. Nephrol. 2010, 6, 643–656.
- Belayev, L.Y.; Palevsky, P.M. The link between acute kidney injury and chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2014, 23, 149–154.
- Venkatachalam, M.A.; Weinberg, J.M.; Kriz, W.; Bidani, A.K. Failed Tubule Recovery, AKI-CKD Transition, and Kidney Disease Progression. J. Am. Soc. Nephrol. 2015, 26, 1765–1776.
- Chevalier, R.L.; Forbes, M.S.; Thornhill, B.A. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 2009, 75, 1145–1152.
- Li, L.; Zepeda-Orozco, D.; Black, R.; Lin, F. Autophagy is a component of epithelial cell fate in obstructive uropathy. Am. J. Pathol. 2010, 176, 1767–1778.
- Kim, W.Y.; Nam, S.A.; Song, H.C.; Ko, J.S.; Park, S.H.; Kim, H.L.; Choi, E.J.; Kim, Y.S.; Kim, J.; Kim, Y.K. The role of autophagy in unilateral ureteral obstruction rat model. Nephrology 2012, 17, 148–159.
- Kim, S.I.; Na, H.J.; Ding, Y.; Wang, Z.; Lee, S.J.; Choi, M.E. Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-β1. J. Biol. Chem. 2012, 287, 11677–11688.
- Ding, Y.; Kim, S.; Lee, S.Y.; Koo, J.K.; Wang, Z.; Choi, M.E. Autophagy regulates TGF-β expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J. Am. Soc. Nephrol. 2014, 25, 2835–2846.
- Nam, S.A.; Kim, W.-Y.; Kim, J.W.; Park, S.H.; Kim, H.L.; Lee, M.-S.; Komatsu, M.; Ha, H.; Lim, J.H.; Park, C.W.; et al. Autophagy attenuates tubulointerstital fibrosis through regulating transforming growth factor-β and NLRP3 inflammasome signaling pathway. Cell Death Dis. 2019, 10, 78.
- Li, H.; Peng, X.; Wang, Y.; Cao, S.; Xiong, L.; Fan, J.; Wang, Y.; Zhuang, S.; Yu, X.; Mao, H. Atg5-mediated autophagy deficiency in proximal tubules promotes cell cycle G2/M arrest and renal fibrosis. Autophagy 2016, 12, 1472–1486.
- Peng, X.; Wang, Y.; Li, H.; Fan, J.; Shen, J.; Yu, X.; Zhou, Y.; Mao, H. ATG5-mediated autophagy suppresses NF-κB signaling to limit epithelial inflammatory response to kidney injury. Cell Death Dis. 2019, 10, 253.
- Bhatia, D.; Chung, K.P.; Nakahira, K.; Patino, E.; Rice, M.C.; Torres, L.K.; Muthukumar, T.; Choi, A.M.; Akchurin, O.M.; Choi, M.E. Mitophagy dependent macrophage reprogramming protects against kidney fibrosis. JCI Insight 2019.
- Kawaoka, K.; Doi, S.; Nakashima, A.; Yamada, K.; Ueno, T.; Doi, T.; Masaki, T. Valproic acid attenuates renal fibrosis through the induction of autophagy. Clin. Exp. Nephrol. 2017, 21, 771–780.
- Nakamura, S.; Oba, M.; Suzuki, M.; Takahashi, A.; Yamamuro, T.; Fujiwara, M.; Ikenaka, K.; Minami, S.; Tabata, N.; Yamamoto, K.; et al. Suppression of autophagic activity by Rubicon is a signature of aging. Nat. Commun. 2019, 10, 847.
- Zhang, Y.; Zhao, S.; Wu, D.; Liu, X.; Shi, M.; Wang, Y.; Zhang, F.; Ding, J.; Xiao, Y.; Guo, B. MicroRNA-22 Promotes Renal Tubulointerstitial Fibrosis by Targeting PTEN and Suppressing Autophagy in Diabetic Nephropathy. J. Diabetes Res. 2018, 2018, 4728645.
- Li, X.-Y.; Wang, S.-S.; Han, Z.; Han, F.; Chang, Y.-P.; Yang, Y.; Xue, M.; Sun, B.; Chen, L.-M. Triptolide Restores Autophagy to Alleviate Diabetic Renal Fibrosis through the miR-141-3p/PTEN/Akt/mTOR Pathway. Mol. Ther. Nucleic Acids 2017, 9, 48–56.
- Shen, Q.; Bi, X.; Ling, L.; Ding, W. 1,25-Dihydroxyvitamin D3 Attenuates Angiotensin II-Induced Renal Injury by Inhibiting Mitochondrial Dysfunction and Autophagy. Cell. Physiol. Biochem. 2018, 51, 1751–1762.
- Tsai, H.C.; Chang, F.P.; Li, T.H.; Liu, C.W.; Huang, C.C.; Huang, S.F.; Yang, Y.Y.; Lee, K.C.; Hsieh, Y.C.; Wang, Y.W.; et al. Elafibranor Inhibits Chronic Kidney Disease Progression in NASH Mice. BioMed Res. Int. 2019, 2019, 6740616.
- Song, Y.; Tao, Q.; Yu, L.; Li, L.; Bai, T.; Song, X.; Hu, H.; Li, Y.; Tan, X. Activation of autophagy contributes to the renoprotective effect of postconditioning on acute kidney injury and renal fibrosis. Biochem. Biophys. Res. Commun. 2018, 504, 641–646.
- Kudo, A.; Kii, I. Periostin function in communication with extracellular matrices. J. Cell Commun. Signal. 2018, 12, 301–308.
- Bian, X.; Bai, Y.; Su, X.; Zhao, G.; Sun, G.; Li, D. Knockdown of periostin attenuates 5/6 nephrectomy-induced intrarenal renin-angiotensin system activation, fibrosis, and inflammation in rats. J. Cell Physiol. 2019, 234, 22857–22873.
- Livingston, M.J.; Ding, H.F.; Huang, S.; Hill, J.A.; Yin, X.M.; Dong, Z. Persistent activation of autophagy in kidney tubular cells promotes renal interstitial fibrosis during unilateral ureteral obstruction. Autophagy 2016, 12, 976–998.
- Khan, I.A.; Nasiruddin, M.; Haque, S.F.; Khan, R.A. Evaluation of Rhubarb Supplementation in Stages 3 and 4 of Chronic Kidney Disease: A Randomized Clinical Trial. Int. J. Chronic Dis. 2014, 2014, 789340.
- Xue, X.; Ren, J.; Sun, X.; Gui, Y.; Feng, Y.; Shu, B.; Wei, W.; Lu, Q.; Liang, Y.; He, W.; et al. Protein kinase Cα drives fibroblast activation and kidney fibrosis by stimulating autophagic flux. J. Biol. Chem. 2018, 293, 11119–11130.
- Tu, Y.; Gu, L.; Chen, D.; Wu, W.; Liu, H.; Hu, H.; Wan, Y.; Sun, W. Rhein Inhibits Autophagy in Rat Renal Tubular Cells by Regulation of AMPK/mTOR Signaling. Sci. Rep. 2017, 7, 43790.