 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Chi-Ting Su | + 2923 word(s) | 2923 | 2021-05-28 05:08:56 | | | |
| 2 | Peter Tang | Meta information modification | 2923 | 2021-06-07 05:31:30 | | |
Video Upload Options
Latent transforming growth factor β (TGFβ)-binding protein (LTBP) 4, a member of the LTBP family, shows structural homology with fibrillins. Both these protein types are characterized by calcium-binding epidermal growth factor-like repeats interspersed with 8-cysteine domains. Based on its domain composition and distribution, LTBP4 is thought to adopt an extended structure, facilitating the linear deposition of tropoelastin onto microfibrils. In humans, mutations in LTBP4 result in autosomal recessive cutis laxa type 1C, characterized by redundant skin, pulmonary emphysema, and valvular heart disease. LTBP4 is an essential regulator of TGFβ signaling and is related to development, immunity, injury repair, and diseases, playing a central role in regulating inflammation, fibrosis, and cancer progression.
1. Introduction
2. Biological Implications
2.1. LTBP4 Structure and Isoforms
2.2. TGFβ-Related Signaling
2.3. Non-TGFβ-Dependent Functions
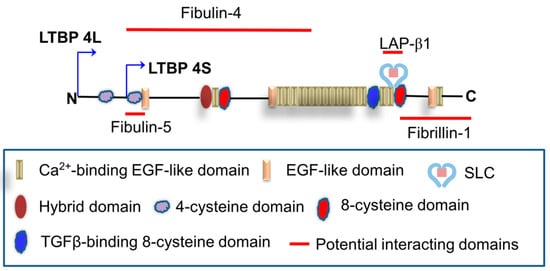
2.4. LTBP4 versus Other LTBP Family Members
3. Clinical Disorders
3.1. Hereditary Diseases
3.1.1. LTBP4-Related Cutis Laxa
3.1.2. Neuromuscular Disorders
3.2. Acquired Complex Diseases
3.2.1. Fibrosis-Related Disorders
3.2.2. Cancers
3.2.3. Pulmonary Disorders
3.2.4. Cardiovascular Disorders
References
- Jensen, S.A.; Robertson, I.B.; Handford, P.A. Dissecting the fibrillin microfibril: Structural insights into organization and function. Structure 2012, 20, 215–225.
- Robertson, I.B.; Horiguchi, M.; Zilberberg, L.; Dabovic, B.; Hadjiolova, K.; Rifkin, D.B. Latent TGF-β-binding proteins. Matrix Biol. 2015, 47, 44–53.
- Koli, K.; Saharinen, J.; Hyytiainen, M.; Penttinen, C.; Keski-Oja, J. Latency, activation, and binding proteins of TGF-β. Microsc. Res Tech. 2001, 52, 354–362.
- Saharinen, J.; Keski-Oja, J. Specific sequence motif of 8-Cys repeats of TGF-β binding proteins, LTBPs, creates a hydrophobic interaction surface for binding of small latent TGF-β. Mol. Biol. Cell 2000, 11, 2691–2704.
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322.
- Xavier, S.; Sahu, R.K.; Landes, S.G.; Yu, J.; Taylor, R.P.; Ayyadevara, S.; Megyesi, J.; Stallcup, W.B.; Duffield, J.S.; Reis, E.S.; et al. Pericytes and immune cells contribute to complement activation in tubulointerstitial fibrosis. Am. J. Physiol. Renal. Physiol. 2017, 312, F516–F532.
- Lack, J.; O’Leary, J.M.; Knott, V.; Yuan, X.; Rifkin, D.B.; Handford, P.A.; Downing, A.K. Solution structure of the third TB domain from LTBP1 provides insight into assembly of the large latent complex that sequesters latent TGF-β. J. Mol. Biol. 2003, 334, 281–291.
- Saharinen, J.; Hyytiainen, M.; Taipale, J.; Keski-Oja, J. Latent transforming growth factor-β binding proteins (LTBPs)—structural extracellular matrix proteins for targeting TGF-β action. Cytokine Growth Factor Rev. 1999, 10, 99–117.
- Yuan, X.; Downing, A.K.; Knott, V.; Handford, P.A. Solution structure of the transforming growth factor β-binding protein-like module, a domain associated with matrix fibrils. EMBO J. 1997, 16, 6659–6666.
- Todorovic, V.; Rifkin, D.B. LTBPs, more than just an escort service. J. Cell Biochem. 2012, 113, 410–418.
- Saharinen, J.; Taipale, J.; Monni, O.; Keski-Oja, J. Identification and characterization of a new latent transforming growth factor-β-binding protein, LTBP-4. J. Biol. Chem. 1998, 273, 18459–18469.
- Kanzaki, T.; Olofsson, A.; Moren, A.; Wernstedt, C.; Hellman, U.; Miyazono, K.; Claesson-Welsh, L.; Heldin, C.H. TGF-β 1 binding protein: A component of the large latent complex of TGF-β 1 with multiple repeat sequences. Cell 1990, 61, 1051–1061.
- Kantola, A.K.; Ryynanen, M.J.; Lhota, F.; Keski-Oja, J.; Koli, K. Independent regulation of short and long forms of latent TGF-β binding protein (LTBP)-4 in cultured fibroblasts and human tissues. J. Cell Physiol. 2010, 223, 727–736.
- Hinck, A.P.; Mueller, T.D.; Springer, T.A. Structural Biology and Evolution of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2016, 8.
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-β family signaling. Sci. Signal 2019, 12.
- Miyazawa, K.; Miyazono, K. Regulation of TGF-β Family Signaling by Inhibitory Smads. Cold Spring Harb. Perspect. Biol. 2017, 9.
- Lodyga, M.; Hinz, B. TGF-beta1—A truly transforming growth factor in fibrosis and immunity. Semin. Cell Dev. Biol. 2020, 101, 123–139.
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-β Family. Cold Spring Harb. Perspect Biol. 2017, 9.
- David, C.J.; Massague, J. Contextual determinants of TGFbeta action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435.
- Zhang, Y.E. Mechanistic insight into contextual TGF-β signaling. Curr. Opin. Cell Biol. 2018, 51, 1–7.
- Luo, K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect Biol. 2017, 9.
- Ayyaz, A.; Attisano, L.; Wrana, J.L. Recent advances in understanding contextual TGFbeta signaling. F1000Res 2017, 6, 749.
- Yu, Y.; Feng, X.H. TGF-β signaling in cell fate control and cancer. Curr. Opin. Cell Biol. 2019, 61, 56–63.
- Isogai, Z.; Ono, R.N.; Ushiro, S.; Keene, D.R.; Chen, Y.; Mazzieri, R.; Charbonneau, N.L.; Reinhardt, D.P.; Rifkin, D.B.; Sakai, L.Y. Latent transforming growth factor β-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J. Biol. Chem. 2003, 278, 2750–2757.
- Noda, K.; Dabovic, B.; Takagi, K.; Inoue, T.; Horiguchi, M.; Hirai, M.; Fujikawa, Y.; Akama, T.O.; Kusumoto, K.; Zilberberg, L.; et al. Latent TGF-β binding protein 4 promotes elastic fiber assembly by interacting with fibulin-5. Proc. Natl. Acad. Sci. USA 2013, 110, 2852–2857.
- Dabovic, B.; Robertson, I.B.; Zilberberg, L.; Vassallo, M.; Davis, E.C.; Rifkin, D.B. Function of latent TGFbeta binding protein 4 and fibulin 5 in elastogenesis and lung development. J. Cell Physiol. 2015, 230, 226–236.
- Bultmann-Mellin, I.; Conradi, A.; Maul, A.C.; Dinger, K.; Wempe, F.; Wohl, A.P.; Imhof, T.; Wunderlich, F.T.; Bunck, A.C.; Nakamura, T.; et al. Modeling autosomal recessive cutis laxa type 1C in mice reveals distinct functions for Ltbp-4 isoforms. Dis. Model Mech. 2015, 8, 403–415.
- Sengle, G.; Sakai, L.Y. The fibrillin microfibril scaffold: A niche for growth factors and mechanosensation? Matrix Biol. 2015, 47, 3–12.
- Robertson, I.B.; Rifkin, D.B. Regulation of the Bioavailability of TGF-β and TGF-β-Related Proteins. Cold Spring Harb Perspect Biol 2016, 8.
- Fujikawa, Y.; Yoshida, H.; Inoue, T.; Ohbayashi, T.; Noda, K.; von Melchner, H.; Iwasaka, T.; Shiojima, I.; Akama, T.O.; Nakamura, T. Latent TGF-β binding protein 2 and 4 have essential overlapping functions in microfibril development. Sci. Rep. 2017, 7, 43714.
- Todorovic, V.; Frendewey, D.; Gutstein, D.E.; Chen, Y.; Freyer, L.; Finnegan, E.; Liu, F.; Murphy, A.; Valenzuela, D.; Yancopoulos, G.; et al. Long form of latent TGF-β binding protein 1 (Ltbp1L) is essential for cardiac outflow tract septation and remodeling. Development 2007, 134, 3723–3732.
- Narooie-Nejad, M.; Paylakhi, S.H.; Shojaee, S.; Fazlali, Z.; Rezaei Kanavi, M.; Nilforushan, N.; Yazdani, S.; Babrzadeh, F.; Suri, F.; Ronaghi, M.; et al. Loss of function mutations in the gene encoding latent transforming growth factor β binding protein 2, LTBP2, cause primary congenital glaucoma. Hum. Mol. Genet. 2009, 18, 3969–3977.
- Ali, M.; McKibbin, M.; Booth, A.; Parry, D.A.; Jain, P.; Riazuddin, S.A.; Hejtmancik, J.F.; Khan, S.N.; Firasat, S.; Shires, M.; et al. Null mutations in LTBP2 cause primary congenital glaucoma. Am. J. Hum. Genet. 2009, 84, 664–671.
- Morkmued, S.; Hemmerle, J.; Mathieu, E.; Laugel-Haushalter, V.; Dabovic, B.; Rifkin, D.B.; Dolle, P.; Niederreither, K.; Bloch-Zupan, A. Enamel and dental anomalies in latent-transforming growth factor β-binding protein 3 mutant mice. Eur. J. Oral. Sci. 2017, 125, 8–17.
- Dabovic, B.; Chen, Y.; Colarossi, C.; Zambuto, L.; Obata, H.; Rifkin, D.B. Bone defects in latent TGF-β binding protein (Ltbp)-3 null mice; a role for Ltbp in TGF-β presentation. J. Endocrinol. 2002, 175, 129–141.
- Dabovic, B.; Levasseur, R.; Zambuto, L.; Chen, Y.; Karsenty, G.; Rifkin, D.B. Osteopetrosis-like phenotype in latent TGF-β binding protein 3 deficient mice. Bone 2005, 37, 25–31.
- Urban, Z.; Hucthagowder, V.; Schurmann, N.; Todorovic, V.; Zilberberg, L.; Choi, J.; Sens, C.; Brown, C.W.; Clark, R.D.; Holland, K.E.; et al. Mutations in LTBP4 cause a syndrome of impaired pulmonary, gastrointestinal, genitourinary, musculoskeletal, and dermal development. Am. J. Hum. Genet. 2009, 85, 593–605.
- Callewaert, B.; Su, C.T.; Van Damme, T.; Vlummens, P.; Malfait, F.; Vanakker, O.; Schulz, B.; Mac Neal, M.; Davis, E.C.; Lee, J.G.; et al. Comprehensive clinical and molecular analysis of 12 families with type 1 recessive cutis laxa. Hum. Mutat. 2013, 34, 111–121.
- Su, C.T.; Huang, J.W.; Chiang, C.K.; Lawrence, E.C.; Levine, K.L.; Dabovic, B.; Jung, C.; Davis, E.C.; Madan-Khetarpal, S.; Urban, Z. Latent transforming growth factor binding protein 4 regulates transforming growth factor β receptor stability. Hum. Mol. Genet. 2015, 24, 4024–4036.
- Ritelli, M.; Cammarata-Scalisi, F.; Cinquina, V.; Colombi, M. Clinical and molecular characterization of an 18-month-old infant with autosomal recessive cutis laxa type 1C due to a novel LTBP4 pathogenic variant, and literature review. Mol. Genet. Genom. Med. 2019, 7, e00735.
- Heinz, A. Elastic fibers during aging and disease. Ageing Res. Rev. 2021, 66, 101255.
- Sterner-Kock, A.; Thorey, I.S.; Koli, K.; Wempe, F.; Otte, J.; Bangsow, T.; Kuhlmeier, K.; Kirchner, T.; Jin, S.; Keski-Oja, J.; et al. Disruption of the gene encoding the latent transforming growth factor-β binding protein 4 (LTBP-4) causes abnormal lung development, cardiomyopathy, and colorectal cancer. Genes Dev. 2002, 16, 2264–2273.
- Dabovic, B.; Chen, Y.; Choi, J.; Vassallo, M.; Dietz, H.C.; Ramirez, F.; von Melchner, H.; Davis, E.C.; Rifkin, D.B. Dual functions for LTBP in lung development: LTBP-4 independently modulates elastogenesis and TGF-β activity. J. Cell Physiol. 2009, 219, 14–22.
- Bultmann-Mellin, I.; Dinger, K.; Debuschewitz, C.; Loewe, K.M.A.; Melcher, Y.; Plum, M.T.W.; Appel, S.; Rappl, G.; Willenborg, S.; Schauss, A.C.; et al. Role of LTBP4 in alveolarization, angiogenesis, and fibrosis in lungs. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 313, L687–L698.
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928.
- Bello, L.; Pegoraro, E. The “Usual Suspects”: Genes for Inflammation, Fibrosis, Regeneration, and Muscle Strength Modify Duchenne Muscular Dystrophy. J. Clin. Med. 2019, 8, 649.
- Chen, Y.; Ali, T.; Todorovic, V.; O’Leary, J.M.; Kristina Downing, A.; Rifkin, D.B. Amino acid requirements for formation of the TGF-β-latent TGF-β binding protein complexes. J. Mol. Biol. 2005, 345, 175–186.
- Flanigan, K.M.; Ceco, E.; Lamar, K.M.; Kaminoh, Y.; Dunn, D.M.; Mendell, J.R.; King, W.M.; Pestronk, A.; Florence, J.M.; Mathews, K.D.; et al. LTBP4 genotype predicts age of ambulatory loss in Duchenne muscular dystrophy. Ann. Neurol. 2013, 73, 481–488.
- Lamar, K.M.; Miller, T.; Dellefave-Castillo, L.; McNally, E.M. Genotype-Specific Interaction of Latent TGFbeta Binding Protein 4 with TGFbeta. PLoS ONE 2016, 11, e0150358.
- Swaggart, K.A.; McNally, E.M. Modifiers of heart and muscle function: Where genetics meets physiology. Exp. Physiol. 2014, 99, 621–626.
- Lu, J.; Liu, Q.; Wang, L.; Tu, W.; Chu, H.; Ding, W.; Jiang, S.; Ma, Y.; Shi, X.; Pu, W.; et al. Increased expression of latent TGF-β-binding protein 4 affects the fibrotic process in scleroderma by TGF-β/SMAD signaling. Lab. Invest. 2017, 97, 591–601.
- Groblewska, M.; Siewko, M.; Mroczko, B.; Szmitkowski, M. The role of matrix metalloproteinases (MMPs) and their inhibitors (TIMPs) in the development of esophageal cancer. Folia Histochem. Cytobiol. 2012, 50, 12–19.
- Dallas, S.L.; Miyazono, K.; Skerry, T.M.; Mundy, G.R.; Bonewald, L.F. Dual role for the latent transforming growth factor-β binding protein in storage of latent TGF-β in the extracellular matrix and as a structural matrix protein. J. Cell Biol. 1995, 131, 539–549.
- Koli, K.; Wempe, F.; Sterner-Kock, A.; Kantola, A.; Komor, M.; Hofmann, W.K.; von Melchner, H.; Keski-Oja, J. Disruption of LTBP-4 function reduces TGF-β activation and enhances BMP-4 signaling in the lung. J. Cell Biol. 2004, 167, 123–133.
- Kong, F.F.; Zhu, Y.L.; Yuan, H.H.; Wang, J.Y.; Zhao, M.; Gong, X.D.; Liu, F.; Zhang, W.Y.; Wang, C.R.; Jiang, B. FOXM1 regulated by ERK pathway mediates TGF-beta1-induced EMT in NSCLC. Oncol. Res. 2014, 22, 29–37.
- Wang, X.; Zhang, Y.; Nilsson, C.L.; Berven, F.S.; Andren, P.E.; Carlsohn, E.; Horvatovich, P.; Malm, J.; Fuentes, M.; Vegvari, A.; et al. Association of chromosome 19 to lung cancer genotypes and phenotypes. Cancer Metastasis Rev. 2015, 34, 217–226.
- Kretschmer, C.; Conradi, A.; Kemmner, W.; Sterner-Kock, A. Latent transforming growth factor binding protein 4 (LTBP4) is downregulated in mouse and human DCIS and mammary carcinomas. Cell Oncol. 2011, 34, 419–434.
- Mauel, S.; Kruse, B.; Etschmann, B.; von der Schulenburg, A.G.; Schaerig, M.; Stovesand, K.; Wilcken, B.; Sterner-Kock, A. Latent transforming growth factor binding protein 4 (LTBP-4) is downregulated in human mammary adenocarcinomas in vitro and in vivo. APMIS 2007, 115, 687–700.
- Klopfleisch, R.; Schutze, M.; Gruber, A.D. Downregulation of transforming growth factor β (TGFbeta) and latent TGFbeta binding protein (LTBP)-4 expression in late stage canine mammary tumours. Vet. J. 2010, 186, 379–384.
- Bultmann, I.; Conradi, A.; Kretschmer, C.; Sterner-Kock, A. Latent transforming growth factor β-binding protein 4 is downregulated in esophageal cancer via promoter methylation. PLoS ONE 2013, 8, e65614.
- Yang, X.; Ye, X.; Zhang, L.; Zhang, X.; Shu, P. Disruption of LTBP4 Induced Activated TGFbeta1, Immunosuppression Signal and Promoted Pulmonary Metastasis in Hepatocellular Carcinoma. Onco. Targets Ther. 2020, 13, 7007–7017.
- Wee, I.; Syn, N.; Sethi, G.; Goh, B.C.; Wang, L. Role of tumor-derived exosomes in cancer metastasis. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 12–19.
- Shin, K.M.; Hong, M.J.; Lee, S.Y.; Jin, C.C.; Baek, S.A.; Lee, J.H.; Choi, J.E.; Kang, H.G.; Lee, W.K.; Seok, Y.; et al. Regulatory variants in cancer-related pathway genes predict survival of patients with surgically resected non-small cell lung cancer. Gene 2018, 646, 56–63.
- Tomasovic, A.; Kurrle, N.; Wempe, F.; De-Zolt, S.; Scheibe, S.; Koli, K.; Serchinger, M.; Schnutgen, F.; Surun, D.; Sterner-Kock, A.; et al. Ltbp4 regulates Pdgfrbeta expression via TGFbeta-dependent modulation of Nrf2 transcription factor function. Matrix Biol. 2017, 59, 109–120.
- Zhou, Y.; Koli, K.; Hagood, J.S.; Miao, M.; Mavalli, M.; Rifkin, D.B.; Murphy-Ullrich, J.E. Latent transforming growth factor-β-binding protein-4 regulates transforming growth factor-beta1 bioavailability for activation by fibrogenic lung fibroblasts in response to bleomycin. Am. J. Pathol. 2009, 174, 21–33.
- Ryan, D.M.; Vincent, T.L.; Salit, J.; Walters, M.S.; Agosto-Perez, F.; Shaykhiev, R.; Strulovici-Barel, Y.; Downey, R.J.; Buro-Auriemma, L.J.; Staudt, M.R.; et al. Smoking dysregulates the human airway basal cell transcriptome at COPD risk locus 19q13.2. PLoS ONE 2014, 9, e88051.
- Hersh, C.P.; Demeo, D.L.; Lazarus, R.; Celedon, J.C.; Raby, B.A.; Benditt, J.O.; Criner, G.; Make, B.; Martinez, F.J.; Scanlon, P.D.; et al. Genetic association analysis of functional impairment in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2006, 173, 977–984.
- Rocchiccioli, S.; Cecchettini, A.; Panesi, P.; Farneti, P.A.; Mariani, M.; Ucciferri, N.; Citti, L.; Andreassi, M.G.; Foffa, I. Hypothesis-free secretome analysis of thoracic aortic aneurysm reinforces the central role of TGF-β cascade in patients with bicuspid aortic valve. J. Cardiol. 2017, 69, 570–576.
- Thompson, A.R.; Cooper, J.A.; Jones, G.T.; Drenos, F.; van Bockxmeer, F.M.; Biros, E.; Walker, P.J.; van Rij, A.M.; Golledge, J.; Norman, P.E.; et al. Assessment of the association between genetic polymorphisms in transforming growth factor β, and its binding protein (LTBP), and the presence, and expansion, of Abdominal Aortic Aneurysm. Atherosclerosis 2010, 209, 367–373.
- Hafez, H.; Druce, P.S.; Ashton, H.A. Abdominal aortic aneurysm development in men following a “normal” aortic ultrasound scan. Eur. J. Vasc. Endovasc. Surg. 2008, 36, 553–558.
- Boileau, A.; Lalem, T.; Vausort, M.; Zhang, L.; Devaux, Y.; Cardiolinc, n. A 3-gene panel improves the prediction of left ventricular dysfunction after acute myocardial infarction. Int. J. Cardiol. 2018, 254, 28–35.
- D’Amario, D.; Vergallo, R.; Crea, F. Predicting the future after acute myocardial infarction: A gaze into the crystal ball of gene expression profile. Int. J. Cardiol. 2018, 254, 47–48.
- Colige, A.; Monseur, C.; Crawley, J.T.B.; Santamaria, S.; de Groot, R. Proteomic discovery of substrates of the cardiovascular protease ADAMTS7. J. Biol. Chem. 2019, 294, 8037–8045.
- Arroyo, A.G.; Andres, V. ADAMTS7 in cardiovascular disease: From bedside to bench and back again? Circulation 2015, 131, 1156–1159.

