 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Grace Hope Daoud | + 5921 word(s) | 5921 | 2021-05-20 05:38:03 | | | |
| 2 | Conner Chen | Meta information modification | 5921 | 2021-06-06 13:35:28 | | |
Video Upload Options
Breast cancer is one of the most prevalent forms of cancer globally and is among the leading causes of death in women. Its heterogenic nature is a result of the involvement of numerous aberrant genes that contribute to the multi-step pathway of tumorigenesis. Despite the fact that several disease-causing mutations have been identified, therapy is often aimed at alleviating symptoms rather than rectifying the mutation in the DNA sequence. The Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 is a groundbreaking tool that is being utilized for the identification and validation of genomic targets bearing tumorigenic potential. CRISPR/Cas9 supersedes its gene-editing predecessors through its unparalleled simplicity, efficiency and affordability.
1. CRISPR/Cas9 System
A major breakthrough in the field of genomics is the development of the CRISPR/Cas9 technology that has revolutionized gene editing in the 21st century. CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) was first identified in Escherichia Coli in 1987 as a group of repeated fragments comprised of 29 nucleotides that are separated by fragments of 32 nucleotides of unique varied sequence [1]. This was shown to play a role in multiple cellular processes including thermal adaptation [2], DNA repair [3] and chromosomal rearrangements [4]. In addition, a comparable 24 to 40 nucleotide short palindromic repeat sequence interspaced by a 20 to 58 varied nucleotide sequence was later identified in multiple species of bacteria and archaea, such as in Streptococcus pyogenes (S.pyogenes), Mycobacterium tuberculosis and Haloferax Mediterranean [5][6]. In 2005, researchers elucidated the homology between the short spacer fragments found on the CRISPR locus and the DNA of prokaryotic invading pathogens. Research over the years showed that CRISPR evolved with time as an adaptive immune system, protecting bacteria and archaea from foreign DNA invaders such as viruses and plasmids [7].
The CRISPR/Cas systems are grouped into 2 classes, 6 types, and 33 subtypes indicated by the involvement of the different Cas proteins within the CRISPR framework that either target DNA, RNA, or both [8][9]. The classification is summarized in Table 1 [10].
Table 1. Classification of the CRISPR/Cas Systems.
| CRIPSR/Cas Systems | ||||||
|---|---|---|---|---|---|---|
| Class | 1 | 2 | ||||
| Protein type | Multiplex | Single | ||||
| Type | I | III | IV | II | V | VI |
| Corresponding Cas protein |
Cas 3 | Cas 10 | Cas 8 | Cas 9 | Cas 12a, Cas 12c, Cas 13a | Cas 13b, Cas 13c |
In 2013, scientists proposed the development of a targeted genome editing tool using the CRISPR/Cas9 technology found in S. pyogenes [11]. Specifically, the class 2 type II subgroup found in this species is most extensively employed for genome editing due to its simplicity necessitating merely a single Cas protein, the endonuclease protein Cas9, along with 2 RNA components, CRISPR RNA (crRNA) and trans-activating CRISPR RNA (tracrRNA). The CRISPR/Cas9 system in S. pyogenes (SpCas9) was further simplified to constitute two components, the Cas9 protein and a single guide RNA (sgRNA) through the hybridization of crRNA and tracrRNA, enabling the manipulation of the eukaryotic genome [11][12][13]. Correspondingly, the immunity provided by the CRISPR/Cas9 system can be characterized into three phases: (1) integration of phage or plasmid DNA into the CRISPR array, (2) CRISPR locus transcription to form pre-crRNA and maturation into crRNA and formation of tracrRNA, and (3) DNA manipulation [14].
The initial phase of CRISPR/Cas9 activity is the integration of short sequences of phage or plasmid DNA, termed protospacer, into the host genome which serves as a cellular memory of past infections. This enables prokaryotes to distinguish subsequent infections by these invaders as foreign, leading to silencing of the alien DNA. The acquired foreign DNA constitutes the varied region, called spacer, found on the CRISPR loci [15]. CRISPR spacer acquisition is mediated by two core proteins, Cas1 and Cas2, which are the only proteins virtually found in almost all the identified CRISPR/Cas systems [16]. A stable complex is formed between these two proteins to initiate the adaptation process; Cas1 possesses endonuclease activity that is necessary for spacer integration while Cas2 seems to play out a non-enzymatic role [17]. It has also been suggested that Cas9 plays a direct role in protospacer acquisition by recruiting Cas1 and Cas2 to potential targets [18].
The second phase begins with the transcription of the CRISPR locus to generate pre-crRNA, a long RNA molecule that contains sequences complementary to those of the spacers and repeats. The tracrRNA is the second RNA molecule needed and is essential for pre-crRNA maturation, it is transcribed from a genomic locus located upstream of the CRISPR locus [19]. The tracrRNA contains a segment that is homologous to the cognate sequence of the CRISPR locus; therefore, is able to bind to the 3′ end of the pre-crRNA forming a double-stranded RNA molecule [20]. Subsequently, the pre-crRNA:tracrRNA double-stranded RNA is cleaved by recruited cellular ribonuclease III (RNase III) responsible for the recognition and cleavage of double-stranded RNA molecules [19]. A second cleavage takes place whereby the 5′ end of the RNA sequence is cut, yielding a mature crRNA:tracrRNA (gRNA) complex ready to associate with a Cas protein, with each individual crRNA fragment containing a unique spacer sequence that is around 20 nucleotides in length [20][21]. The resulting gRNA complex binds to the Cas9 protein, creating a Cas9:gRNA effector complex capable of DNA interference to complete the CRISPR mediated immunity. The Cas9 protein is a dual RNA-guided endonuclease enzyme having a bi-lobed structure, the α-helical recognition (REC) lobe and the nuclease lobe, with the RNA complex situated in between. The latter is comprised of two nuclease domains, an HNH domain responsible for cleaving the complementary DNA strand to crRNA and a RuvC-like domain which cleaves the non-complementary DNA strand. On the other hand, the REC lobe contains an arginine-rich bridge that is essential for RNA interaction and joining the two lobes together [22][23][24]. Once Cas9 is activated by binding to the gRNA complex, it scavenges for any invading nucleic acid sequences that show complementarity to the crRNA. Therefore, CRISPR/Cas9 initiates a double-stranded cleavage at a specified DNA sequence site following base pairing of crRNA to the target site [25] (Figure 1).
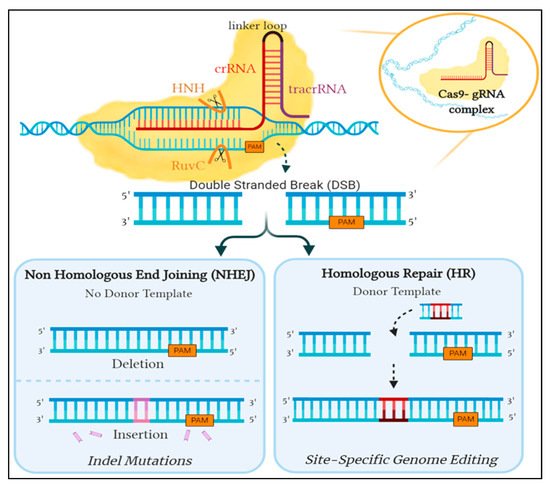
Figure 1. An overview of the repair mechanism associated with induced Cas9 double-stranded DNA break. Cleavage is induced by the binding of Cas9- gRNA complex to its complementary sequence on foreign DNA. In eukaryotes, this is amended by either of two mechanisms: the error-prone Non-Homologous End Joining (NHEJ) or Homologous Repair (HR), which is utilized for genome editing by providing a donor template. Created with Biorender.
However, the prospective target sequence is only valid if a short sequence known as Protospacer Adjacent Motif (PAM) is present directly after the binding location of crRNA. The presence of PAM is the underlying factor that determines preference between self and non-self DNA. Although the CRISPR array contains spacers that are identical to foreign DNA, the CRISPR genome is not affected by its own mechanism as the spacers do not lie immediately next to a PAM sequence [26]. In addition, the PAM sequence identified by Cas9 varies between microorganisms; SpCas9 specifically recognizes 5′-NGG-3′ [25], resulting in a blunt-end double-strand break occurring upstream by three base pairs in the PAM sequence [20]. The guanine dinucleotide [22] of PAM found on the non-complementary strand aids in its recognition by interacting with two crucial arginine residues. Further interactions form a bend in the target DNA assisting in the unwinding of the helical structure which propagates cutting of the intruder DNA [27]. This disruption in the invading pathogen is deemed to be detrimental to its existence and is ultimately what provides protection for the prokaryote.
This guided interference into the DNA sequence inspired researchers to exploit the system with hopes of achieving precise genome editing. Unfortunately, the CRISPR/Cas9 system in prokaryotes utilizes components not inherently present in eukaryotes, prompting the need to optimize the S. pyogenes’ CRISPR/Cas9 system. The modifiable crRNA is merged with the tracrRNa to form sgRNA, which works similarly to the gRNA complex as it guides Cas9 to the target sequence site and triggers the cleavage of both DNA strands. The double-strand DNA breaks induced by Cas9 can then be amended by one of two DNA repair pathways: either the non-homologous end joining (NHEJ) or the homology-directed repair (HDR) [28]. NHEJ ligates the broken ends together, however, this pathway is error-prone and could lead to insertion/deletion (indel) mutations resulting in an ineffective gene. In contrast, HDR uses a neighboring homologous sequence as a template to mend the break. This method can be exploited to potentially introduce targeted edits at a precise location into the DNA sequence by providing a donor template attached to sgRNA for repair [20] (Figure 1).
As the machinery behind the whole CRISPR/Cas9 system relies on the complementarity between crRNA and the target sequence, in addition to the presence of PAM [25], specificity is crucial when generating a sgRNA. In case the desired target is inaccurately outlined, Cas9 can bind and cause an off-target cleave of the sequence, leading to unintended mutations that could be consequential. However, as long as the target sequence is identified, several CRISPR software tools are available to facilitate the design of an optimal sgRNA to achieve precise cleavage with minimal off-target effects [29]. Owing to the versatility in sequencing sgRNAs, attachment of the Cas9:gRNA complex to various sites is plausible. This merits the CRISPR/Cas9 system to reconstruct a multitude of loci concurrently [30].
2. Potential CRISPR/Cas9 Targets
2.1. Genes Involved in Tumorigenesis
Mutations in the genes regulating several cellular processes, including but not limited to cell survival, proliferation, motility and apoptosis, deregulate the gene normal function which, as a result, could promote uncontrolled cellular growth, giving rise to tumor formation). Discussed below are critical genes involved in the pathogenesis of breast cancer with supporting studies employing CRISPR/Cas9.
2.1.1. BRCA1
The BRCA1 gene is expressed in a number of tissues, including ovarian and breast tissue. Acting as a tumor suppressor, it is involved in multiple cellular regulatory pathways including gene transcription regulation, ubiquitination, cell-cycle progression and DNA-damage response; the latter being a pathway in which BRCA1 plays a pivotal role [31]. Such versatility in its role is a result of possessing multiple functional domains that aid in checkpoint regulation, single-strand annealing (SSA), HR and NHEJ [32]. These include the N-terminal RING (Really Interesting New Gene) domain, the BRCT (BRCA1 C-terminal) domain and exon regions 11–13. Site I and Site II are Zn2+ binding loops responsible for stabilizing RING finger structures; containing four cysteine and three cysteine residues with one histidine residue, respectively. Mutations in the cysteine residues result in altered functions such as decreased ubiquitin ligase activity, which has been shown to increase cancer risk [33][34]. The BRCT domain in BRCA1 functions to regulate interactions of phosphoproteins with BRCA1 as well as facilitating non-phosphoprotein interactions with DNA binding. Mutations of a residue would therefore hinder the role of BRCA1. Such is the case of BRCA1 falling into the “similarity trap” (Figure 2) [35][36]. Normally, 53BP1 (p53 binding protein I) has a higher affinity to phosphorylated p53 than BRCA1. Mutated BRCT domains in BRCA1 reverse the affinity between 53BP1 and BRCA1, which then alters p53 function, possibly precipitating cancer [36].
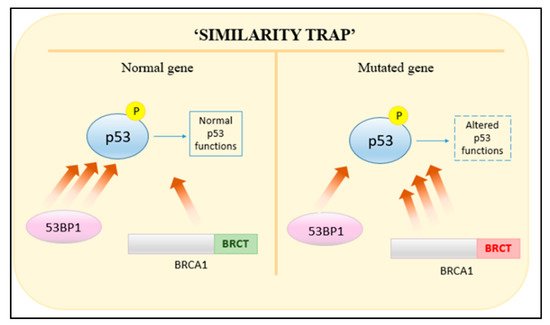
Figure 2. Reversal of affinities between mutated BRCA1 and 53BP1 towards phosphorylated p53.
Furthermore, several variants and non-coding regions are yet to be discovered owing to the complexity within the BRCA1 genomic structure. A study implementing the CRISPR-mediated cytosine Base Editor 3 (BE3) to induce targeted T: A conversions from C: G assessed the functionality of variants of uncertain significance (VUSs) and identified loss-of-function (LOF) variants through high-throughput screens; which can aid in the reclassification of BRCA1 (VUSs). The HAP1 cell line was used to introduce LOF mutations, however prior to that, to demonstrate the feasibility of the experiment, transfection of BRCA1-targeting gRNAs into HAP1-Cas9 cell lines was carried out to derange BRCA1.
Since changes in cell viability due to loss-of-function (LOF) mutations in BRCA1 variants can be used to assess the function of BRCA1, targeted deep sequencing was used to measure mutation frequencies, where the relative indel frequencies decreased substantially with time. BE3-expressing HAP1 (HAP1-BE3) and Cas9-expressing HAP1 (HAP1-Cas9) cell lines were generated with lentiviral particles. After inducing nucleotide substitution mutations by HAP1-BE3 cell lines, a collective gRNA targeting BRCA1 library was designed to accomplish CRISPR-based high-throughput screens [37].
2.1.2. BRCA2
Multiple studies have demonstrated an overlap between cancer outcomes and BRCA1/BRCA2 carriers [38] despite the lack of homology between these genes [39]. The BRCA2 domains are essentially associated with RAD51; a protein related to homologous repair (HR) [40]. DNA repair mechanisms are maintained by BRCA2 through its cyclin-dependent kinase (CDK) interaction with RAD51 to aid in HR—an established role of BRCA2 [41]. In addition, BRCA2 protects nascent strands during replication from degradation [42].
Paradoxically, however, the transmission of replication stress responsible for precancerous lesions to the succeeding cell cycle due to BRCA2 deficiency causes missegregation, as 53GP1 nuclear bodies are formed at the G1 phase. This eventually causes cell inviability as the p53-dependent G1 is seized, as was demonstrated by Feng W. and Jasin M. through CRISPR/Cas9-mediated gene targeting of BRCA2 [43]. These results point out a potential barrier that must be tamed for the commencement of tumorigenesis.
2.1.3. HER2
The Human Epidermal Growth Factor 2 (HER2 or HER2/neu or ERBB2) gene is an oncogene that encodes for the HER2 protein, a membrane receptor tyrosine kinase that is one member of four in the ERBB family (HER1-4) found on breast cells. Amplification of HER2 and the resulting overexpression of its protein is found to be important in the carcinogenesis of the HER2-positive breast cancer subtype [44]. The HER2 signaling pathway, among others, is a major driver of tumor cell proliferation and survival in this breast cancer subtype, and an effective therapeutic target of the monoclonal antibody trastuzumab [45][46]. The HER2 protein, along with the other ERBB family members, consists of three domains: an extracellular, transmembrane and intracellular domain. Once a ligand binds to the extracellular domain, the HER protein will dimerize and result in autophosphorylation of the intracellular domain, which in turn interacts with other signaling molecules that initiate the activation of a variety of downstream signaling pathways involved in cell proliferation, survival and opposes apoptosis. Interestingly, the HER2 protein does not have an associated ligand but rather relies on heterodimerization with any of the other three HER proteins or homodimerization when overexpressed for activation [47]. In addition, heterodimers with the HER2 protein possess the greatest signaling activity, as it has the strongest kinase activity [48].
Targeting of HER2 via CRISPR/Cas9 led to inhibition of cell proliferation and carcinogenesis of breast cancer cells. Exons 5, 10 and 12 of HER 2 were specifically targeted by selective gRNAs, these exons exist in all HER2 isoforms and are responsible for encoding parts of the extracellular domain. Cas9, along with three gRNA, was introduced into HER2+ breast cancer cell lines BT-474 and SKBR-3 and the HER2- breast cancer cell line MCF-7. The co-expression of Cas9 and gRNAs substantially suppressed cell growth in HER2+ cell lines but not in HER2- cell lines, a result that is comparable to that obtained from trastuzumab treatment. In addition, the introduction of Cas9 and gRNAs to a soft agar colony formation assay caused a considerable reduction in colony formation. These results indicate that utilizing CRISPR/Cas9 to target HER2 yields decreased cell proliferation and carcinogenesis, though the effect is limited to HER2+ cell lines [45].
2.1.4. TP53
A widely acknowledged tumor suppressor is the p53 protein, encoded by the TP53 gene. Regrettably, mutations in this gene account for half of the cancer cases, making this the most commonly mutated gene in cancer [49]. In many instances, an amino acid substitution in the p53 protein ensues from one nucleotide mutation in the TP53 gene. These are usually frameshift mutations with the resulting protein possessing non-functional tumor-suppressing and diminished transcriptional activity [50]. Additionally, the mutation stimulates oncogenesis and can affect advanced cancer stages and cancers unresponsive to treatments [51]. Some malignancies, despite possessing the wild-type (WT) TP53 gene, generate multiple cancers with inactive p53 proteins. Post-translational modifications through overexpression of epigenetic regulators in this protein damage its tumor-suppressive properties. Reactivating p53 by inhibiting the contributing epigenetic factors such as methyltransferases and Aurora A kinases could prove to be beneficial towards cancer suppression [52].
However, studying the effect of inducing Cas9 expression in cells, Enache et al. observed p53 pathway activation in Cas9-expressing cell lines. When WT TP53 expressing and TP53 LOF mutated cell lines were compared, the former cell line was most commonly found to have the pathway activated, undeniably because the well-established role of p53 in TP53 LOF cell lines is inactivated. However, not only was there an increase in the number of cells with DNA DSBs upon Cas9 expression but, in addition to other genes, TP53 mutations were observed in Cas9 expressing cell lines. The study also indicated that upon Cas9 expression, cells harnessing the TP53 LOF mutation paradoxically showed increased cell growth; an effect exclusive to TP53 and not in other tumor-suppressing genes, indicating a possible limitation when conducting experiments on TP53 with Cas9 [53]. In this context, it is important to note that findings on targeting TP53 with CRISPR/Cas9 could be extended to other types of cancers knowing that TP53 is a commonly mutated gene in various types of cancer.
2.1.5. TP53PB1
Since the concept of defective DSB repair by the LOF of BRCA1 protein aids in chemotherapy of BRCA1/2 deficient cancers by hyper-sensitizing cells to Poly (ADP-ribose) polymerase (PARP) inhibitors, a study [54] set out to understand the mechanisms by which resistant clones of PARP inhibitors arise, specifically focusing on the loss of 53BP1-encoded by the TP53BP1 gene and its associated co-factors. It is important to note that the chief binding partner of BRCA2, the PALB2 (partner and localizer of BRCA2) gene colocalizes with BRCA2 and aids in its stabilization [55].
It encodes for the PALB2 proteins which bind to BRCA1 and BRCA2 proteins, thereby facilitating the repair of DSBs through homologous recombination [56][57]. Specifically, PALB2 links the BRCA complex (BRCA1-PALB2-BRCA2-RAD51), hence aiding the role of RAD51 in the strand invasion step of homologous recombination [58]. This breast cancer susceptibility gene, therefore, is vital to maintaining the integrity of the genome. Lack or dysfunction thereof would predispose to breast cancer [59]. Nonetheless, the role of 53BP1 in the multiple HR stages was characterized in depleted cells of BRCA1, PALB2 or BRCA2 by short-interfering RNA (siRNA) technology, where HR repair was shown to be strictly inhibited.
However, the depletion of 53BP1 did not restore HR in PALB2 or BRCA2 deficient cells. On the contrary, HR was enhanced in BRCA1/53BP1 depleted cells. To determine whether such disproportional preferences in HR repair could be on account of partial 53BP1 depletion, CRISPR/Cas9 mediated knockout (KO) of the 53BP1 gene was performed, followed by HR assays. PALB2 depletion almost entirely abolished HR both in 53BP1-sufficient and 53BP1-KO cells, indicating that 53PB1 loss reinstates HR in BRCA1 deficient cells, but not in PALB2 deficiency [54].
The aforementioned partial nature of HR depicts that in BRCA1 deficiency, 53PB1 is not proficiently displaced from the chromatin nearby the DSBs [60]. It is still bound to nucleosomes and acknowledged by multiple chromatin-interacting proteins. Loss of 53PB1, on the other hand, exposes its now-vacant nucleosome surfaces for interaction with scarce or even factors avidly binding to the surfaces, such as PALB2.
Briefly, the lack of 53BP1 in BRCA1 deficiency creates a preferable setting for the recruitment of PALB2. Therefore, it was shown that deficient cells of BRCA1/53BP1 led to the accumulation of PALB2 into the damaged DNA foci. Such findings elucidate the role of DNA repair by PALB2 in clinically significant BRCA1/53BP1 deficient cells [54].
2.1.6. MKI67
Ki-67 is a widely used oncogenic biomarker as it is exclusively expressed in proliferating the cells of vertebrates [61]. This nuclear protein, encoded by MKI67, is utilized to grade tumors in histopathology and can be used as a prognostic marker [62]. However, recent studies have invalidated the claims of Ki-67 playing a role in cancer proliferation [58][63][64][65]. Since the proliferation of Ki-67 is controlled by regulators of the cell cycle such as CDKs and B-Myb [64][66][67], Ki-67 in itself is not overexpressed in cancers. Nevertheless, Ki-67 might prove to be essential in carcinogenesis by other means. Its ability to organize heterochromatin is evident by the disruption of nucleoli and centromeres in Ki-67 KOs and the formation of heterochromatin ectopically in its overexpression [64].
Widespread changes in the transcriptome were observed upon the knockout of Ki-67, which indicated that, instead of Ki-67 directly governing specific transcription factors, it interacts with multiple chromatin regulators to give such ‘global transcriptome changes’. This was tested on cell lines simulating TNBC [68][69]. CRISPR/Cas9 mediated gene KO of MKI67 showed no change in the proliferation rates of the tested cell line but rather, the KO caused a genome-wide alteration in the gene expression. In addition, being an intrinsically disordered protein (IDP), the study also concluded that the expression of Ki-67 has an impact on contributing pathways involved in carcinogenesis, ranging from initiation and progression to metastasis. Consequently, the lack of enzymatic activities within the IDP will prove to be an obstacle in the context of finding therapeutic targets. However, inhibiting its effectors and interacting proteins could deem to be of therapeutic value [69]. Similar to TP53, as MKI67 is a proliferation maker in various malignancies, findings on targeting MKI67 by CRISPR/Cas9 can have broader applications that encompass several types of cancers.
2.2. Genes Involved in Metastasis
A major life-threatening problem of cancer cells, particularly those of breast cancer, is their tendency to metastasize to remote tissues across the body, making them responsible for the majority of cancer-related mortalities [70]. The metastatic process begins with the tumor cells departing from their site of origin, followed by intravasation into the bloodstream through the extracellular matrix (ECM) [71][72] where the circulating tumor cells (CTC) work to overcome several cellular obstacles including defensive immune cells. It then attaches to a secondary site, where it has to acclimate for its survival and further develop into a secondary tumor [73][74]. Epithelial-to-Mesenchymal Transition (EMT) is suggested to aid the metastatic process by decreasing the polarity of epithelial cells, yielding an aggressive mesenchymal cell that has the ability to migrate and invade with decreased susceptibility to apoptosis [75][76][77].
2.2.1. MIEN1
Migration and invasion enhancer 1 (MIEN1), until recently named C35 or C17orf37, is a novel breast cancer oncogene that is extensively expressed in all types of breast cancers primarily in HER-2 and luminal B subtypes [78]. MIEN1 is present specifically in the ERBB2 amplicon and is fundamental in regulating the migration and invasion of cancer cells [79]. MIEN1 is involved in several pro-metastatic signaling processes including gene expression through Protein kinase B (Akt) activation [80]. Physiologically, MIEN1 is associated with the formation of filopodia to accelerate cellular motility during tumor dissemination, in addition to controlling cell apoptosis. Despite this, the exact part MIEN1 plays in tumorigenesis and metastasis remains ambiguous. In its C-terminal, MIEN1 constitutes a CAAX motif that acts as a substrate for prenylation, which is the process of covalently adding hydrophobic isoprenoid groups post-translationally to proteins, enhancing their hydrophobicity and accelerating the migration of cells. MIEN1 is prenylated by the enzyme Geranylgeranyltransferase-I (GGTase-I), subsequently promoting protein–inner membrane interactions along with directional migration [81]. The CRISPR/Cas9 tool was employed for precise targeting in MIEN1 gene deletion of MDA-MB-231 breast cancer cell lines where a segment of the gene was knocked using a two sgRNA co-transfection approach to ensure stability. Based on the results, this co-transfection leads to elevated on-target efficiency whereby most of the cell lines demonstrated no expression of the MIEN1 protein. Moreover, the results also inferred no apparent discrepancies in tumor proliferation, morphology or vitality between the MIEN1 KO cells and the original cell lines. The study illustrated the significance of further research into the specifics of MIEN1-mediated oncogenesis in order to substantiate its prospects as a clinical target and biosignature in breast cancer and how CRISPR could be integral in this process [82].
2.2.2. CX3CR1
CX3 chemokine receptor 1 (CX3CR1) is a protein encoded by the CX3CR1 gene and has been implicated in the dissemination of cancer cells and facilitation of cell survival and viability [83]. The only chemokine ligand of CX3CR1 is Fractalkine (FKN or CX3CL1), found expressed as either a transmembrane protein or a soluble protein, where it functions as a chemoattractant [84]. Therefore, the interaction between FKN and CX3CR-expressing CTC is crucial to the precipitation of secondary cancer lesion formation in the skeleton and other soft-tissue organs [83]. Examination of breast cancer specimens of luminal A, luminal B, HER2 and TNBC subtypes all indicated comparable expression of CX3CR1 [85]. Breast cancer patients with bone metastasis show no correlation to breast cancer subtypes, which implies the large role of CX3CR1 in skeletal dissemination [86].
A CRISPR-mediated silencing of CX3CR1 transcription in MDA-MB-231 cell lines led to the ablation of in vitro CX3CR1 protein expression. Upon the introduction of these cells in mice, there was a significant reduction in skeletal and lung metastasis [83]. Comparable results were obtained from FKN knockout transgenic mice (also grafted with MDA-MB-231) indicating that CX3CR1 is directly involved in the lodging of breast cancer cells to the bone [87][88]. Therefore, interfering with the pairing of CX3CR1 with FKN, through the deletion of CX3CR1, can drastically limit the capability of breast cancer circulating tumor cells to disseminate and cause secondary tumors [83], particularly in the bones.
2.2.3. CXCR2
Interleukin 8 (IL-8 or CXCL8) is a chemoattractant cytokine secreted from various cells such as leukocytes, endothelial cells, fibroblasts and malignant tumor cells under certain environmental stressors and takes part in malignant cell migration, proliferation and angiogenesis [89][90]. IL-8 produces an effect upon binding to its receptors, CXC chemokine receptors 1 and 2 (CXCR1 and CXCR2), which are heterodimeric receptors primarily expressed on immune cells such as neutrophils, but may also be present on the surface of various tumor cells [91]. Binding with CXCR2 (encoded by the CXCR2 gene) facilitates cell migration [92] and is therefore reported to play a vital role as a receptor for tumor metastasis [93].
A significant increase in proliferation and migration was observed in MDA-MB-231 cell lines in conditioned media of fibroblasts and macrophages treated by tumor-conditioned media (TCM) of TNBC. This suggests that the migration and proliferation of TNBC cells is enhanced by the crosstalk among TNBC cells and either fibroblasts or macrophages. Several factors were secreted in conditioned media of fibroblasts and conditioned media of macrophages that were treated with tumor-conditioned media of TNBC. However, the secretion and expression of IL-8 was highly upregulated by both fibroblasts and macrophages, suggesting it could be a key factor that fosters TNBC cell proliferation and migration. A CRISPR/Cas9 mediated knockout of CXCR2 in MDA-MB-231 cell lines led to a significant decrease in tumor cell proliferation and migration, compared to the wild-type TNBC cell line. A decrease in metastasis was also observed in the xenograft mouse model. Therefore, these observations indicate that the IL-8-CXCR2 axis is involved in TNBC cell growth and metastasis through crosstalk of TNBC cells, fibroblasts and macrophages [94].
2.2.4. CXCR4 and CXCR7
CXC Motif Chemokine Ligand 12 (CXCL12), otherwise known as Stromal Cell-Derived Factor 1 (SDF1), is a chemokine protein that can regulate cell proliferation, motility and angiogenesis through its interaction with CXC Chemokine Receptor 4 (CXCR4) and CXC Chemokine Receptor 7 (CXCR7) [95]. In addition, it is also highly expressed in organs such as the bone marrow, liver and lungs, allowing simple metastasis of breast cancer cells to such organs [96]. CXCR4 is a transmembrane G-protein coupled receptor encoded by the CXCR4 gene. Attachment of CXCL12 to CXCR4 activates a plethora of downstream signaling pathways resulting in various responses such as cell proliferation, survival, and migration [97]. The CXCL12/CXCR4 axis is said to have a critical role in breast cancer metastasis [95].
In addition to CXCR4, CXCL12 can also bind to CXCR7, a receptor that is overexpressed within the primary tumors vascular system and holds a more elaborate role in breast cancer progression [98][99]. CXCR7 fosters tumor angiogenesis by its influence on cancer cells to secrete vascular endothelial growth factor (VEGF) and promoting metastasis by augmenting cancer cell adhesion to endothelial and fibrin cells [99][100][101]. Overexpression of CXCR7 in breast cancer tissue promotes cell proliferation, invasion and metastasis, particularly to the lungs [101]. Moreover, CXCL12 along with its receptors CXCR4 and CXCR7, are overexpressed in TNBC compared to other subtypes and are correlated to metastasis and poor prognosis [102].
The CRISPR/Cas9 system was utilized to perform single-gene knockout and co-knockout of CXCR4 and CXCR7 genes in the TNBC cell line MDA-MB-231. The single-gene knockout of either CXCR4 or CXCR7 led to no protein expression and caused a significant inhibition of TNBC growth, proliferation, invasion and migration in vitro. However, co-knockout of CXCR4 and CXCR7 caused a more substantial decrease effect as opposed to single-gene knockout, suggesting a synergistic relationship between the two receptors in TNBC progression. These results indicate that CXCL12, CXCR4 and CXCR7 all have an important role in TNBC progression; therefore, co-knockout of both receptors may be a possible therapeutic target in TNBC treatment [96].
2.2.5. MAP3K11
The mitogen-activated protein kinase (MAPK) pathway is involved in the regulation of multiple cellular processes including survival, proliferation, differentiation, motility and apoptosis [103]. This signal transduction is highly regulated by a relay of three protein kinases, MAP kinase kinase kinase (MAP3K), MAP Kinase Kinase (MAP2K) and MAP kinase (MAPK). The activated MAPK proteins can phosphorylate substrates within the cytosol or regulate transcription factors through nuclear translocation. Alteration or overexpression of these MAPK proteins or their upstream regulators results in upregulation of the signal transduction pathway leading to a sustained activation signaling for cancer cells [104].
Mixed-lineage kinase (MLK3) is a MAP3K that has been shown to be overexpressed in TNBC and is critical for its metastasis [105]. In addition, MLK3 is able to activate the activator protein 1 (AP-1) transcription factor—a heterodimer comprised of JUN, FOS, Activating transcription factor (ATF) and MAF that regulates gene expression and controls numerous cellular processes. Furthermore, the FOS-related antigen 1 (FRA1) belonging to the FOS family of proteins is regulated by MLK3 and is found in high amount in TNBC cells and plays relevant roles in tumor cell proliferation and invasion [106]. In addition, FRA1 regulates matrix metalloproteinase (MMP) 1 and 9, which are zinc-dependent endopeptidases involved in the degradation of extracellular matrix protein, aiding in cancer cell invasion and metastasis by remodeling the extracellular matrix [107]. All these factors that facilitate TNBC metastasis are regulated by MLK3 signaling. CRISPR/Cas9 depletion of MLK3 in highly metastatic murine TNBC 4T1 cells led to reduction of FRA-1 and consequently MMP1 and MMP9, which resulted in impairment of tumor cell invasion and migration [108].
2.2.6. TNFRSF11B
Osteoprotegerin (OPG) is a cytokine belonging to the Tumor Necrosis Factor (TNF) superfamily encoded by the TNFRSF11B gene and is broadly known for its pre-emptive role in osteoclastic regulation through the RANK Ligand (RANKL). This ligand is released by osteoblasts and normally binds to the RANK receptors found on the surface of osteoclasts. OPG interacts with RANKL thereby inhibiting the interaction between the ligand and receptor, as a result diminishing both bone reabsorption and osteoclast development [109]. Comprehensive research suggests that OPG is an important tumor modulator and studies on breast cancer cell lines have shown a 40% expression of OPG compared to normal breast cells [110]. During early tumor development, OPG works by interacting with TNF-Related Apoptosis-Inducing Ligand (TRAIL; TNFSF10), another cytokine that is known to promptly induce apoptosis in cell lines. The interaction between OPG and TRAIL blocks the process of apoptosis in tumor cells enhancing their survival [111][112] Gene modifications in OPG as well as its target RANKL has previously been conducted in animal models using the CRISPR/Cas9 system for research pertaining to bone defects [113] and perinatal brain injury [114]. More recently, in vitro studies involving OPG gene KO with the help of CRISPR/Cas9 have also been accomplished in MCF-7 breast cancer cell lines. OPG KO has been shown to inhibit the protein expression of Fatty Acid Synthase (FASN), an essential enzyme that is involved in the fatty acid biosynthetic pathway and is vital for breast cancer existence [115].
2.2.7. UBR5
The Ubiquitin-Proteasome System (UPS) is a crucial regulator of cell signaling and proteostasis essential for cellular processes such as protein catabolism, apoptosis and cell cycle progression. Ubiquitination is a common post-translational modification (PTM) that involves activation, conjugation and ligation through the action of E1 ubiquitin-activating enzymes (E1), E2 ubiquitin-conjugating enzymes (E2) and E3 ubiquitin-ligase (E3), respectively in a sequential manner [116]. E3 Ubiquitin ligase holds a key position within the UPS as it recruits an E2 ubiquitin-conjugating enzyme and catalyzes the ubiquitination of a protein substrate. Such conjugated proteins may take part in oncogenesis [117].
BR5, a member of the E3 ligase family, has been overexpressed in the TNBC subtype unlike luminal A and B subtypes. Consequently, there may be a correlation between advanced clinical cancer stages with the expression of UBR5. Nonetheless, a successful knockout of the UBR5 in 4T1 and B16 cells was attained and showed characteristic morphological changes adapted by the epithelial cells into the mesenchymal shape that aids in the EMT. On the contrary, E-cadherin expression was entirely abolished in 4T1 cells devoid of UBR5. Since this protein plays a role in acquiring the invasive properties during EMT and assists in the advancement of late-stage breast cancer, its loss impairs the Mesenchymal-Epithelial Transition (MET) and hence the ability to settle into secondary organs. Additionally, decreased angiogenesis was observed in histological findings of the 4T1/ubr5-/- tumor 8 days post its inoculation. In short, the UBR5 gene plays a role in the enhancement of initial phases of tumor migration and invasion while limiting the later steps of metastatic colonization [118].
The different stages involved in the carcinogenesis of breast cancer with respect to some of the contributing genes are illustrated in Figure 3, and an overview of the potential therapeutic targets for breast cancer studied in literature employing the CRISPR/Cas9 technology is represented in Table 2.
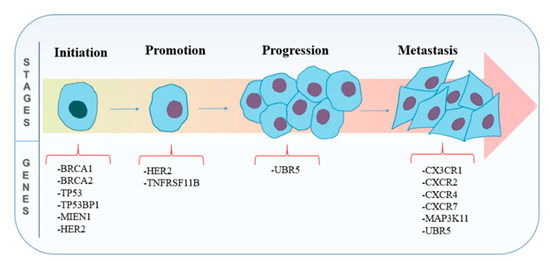
Figure 3. The different stages involved in carcinogenesis of breast cancer with respect to some of the contributing genes.
Table 2. An overview of the potential therapeutic targets for breast cancer studied in literature employing the CRISPR/Cas9 technology.
| Gene | Protein Encoded | Role in Breast Cancer | Associated Subtype of Breast Cancer | CRISPR/Cas9 Application | Results | Ref. |
|---|---|---|---|---|---|---|
| BRCA 1 | BRCA1 | Lacks DSB repair ability | TNBC | Nucleotide substitution through BE | Successful identification of LOF variants | [31][37][119] |
| BRCA 2 | BRCA2 | Lacks DSB repair ability | ER+ and HER2- | KO | Cell inviability | [41][43][119] |
| HER2 | HER2 | Promotes cell proliferation | HER2+ | KO | Suppressed cell proliferation and tumorigenesis | [45] |
| TP53 | p-53 | Deregulates cell cycle | TNBC and/or HER2+ | Cas9 expressed in cell lines | Cas9 induces TP53 mutation | [52][53][120] |
| TP53BP1 | 53BP1 | Lacks DSB repair ability | TNBC | KO | Restoration of HR in BRCA1 and P53BP1 deficient cells | [54][121] |
| MKI67 | Ki-67 | Promotes initiation, progression, metastasis | N/A | KO | Wide-spread transcriptome changes | [61] |
| MIEN1 | MIEN1 protein | Promotes initiation, metastasis | TNBC | KO | No effect on cell viability and tumor proliferation | [82] |
| CX3CR1 | CX3CR1 | Promotes skeletal and soft tissue organ metastasis | Luminal A&B, HER2+, TNBC | Transcription silencing | Reduction in skeletal and lung metastasis | [83] |
| CXCR2 | CXCR2 | Promotes cell proliferation, migration, angiogenesis | TNBC | KO | Suppressed cell proliferation, migration and metastasis | [94] |
| CXCR4 and CXCR7 | CXCR4 and CXCR7 | Promotes cell proliferation, invasion, metastasis | TNBC | Single-gene knockout and co-knockout | Suppressed cell proliferation, invasion and migration | [96] |
| MAP3K11 | MLK 3 | Promotes metastasis | TNBC | KO | Suppressed cell invasion and migration | [108] |
| TNFRSF11B | OPG | Blocks apoptosis | HR+ | KO | Inhibited protein expression of Fatty Acid Synthase | [115] |
| UBR5 | Ubr5 | Aids EMT | TNBC | KO | Induced apoptosis and suppressed metastasis | [118] |
| CDK4 | CDK4 | Cell proliferation | TNBC and HER2+ | KO | Suppressed viability, clono-genicity, migration | [122] |
| MFN2 | MFN2 | Suppress cancer progression via mTOR2/Akt signal inhibition | HR+ | KO | Promotes cell viability, invasion, colony formation | [123] |
| APOBEC3G | APOBEC3 | APOBEC3 induced mutagenesis | HER2+ | KO | Suppressed cell proliferation | [124] |
| MARK4 | MARK4 | Inhibits Hippo signaling leading to cell proliferation | TNBC | KO | Suppressed cell proliferation and migration | [125] |
| MASTL | MASTL | Promotes proliferation | HR+ and TNBC | KO | Suppressed proliferation and tumor growth | [126] |
| MELK | MELK | Tumorigenesis regulator | TNBC | KO | CRISPR/Cas9-Mediated Mutagenesis | [127] |
| MFGE8 | MFGE8 | Mediator of breast cancer tumorigenesis | TNBC | KO | Restored sensitivity to COX-2 selective inhibitor | [128] |
References
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433.
- Riehle, M.M.; Bennett, A.F.; Long, A.D. Genetic architecture of thermal adaptation in Escherichia coli. Proc. Natl. Acad. Sci. USA 2001, 98, 525–530.
- Makarova, K.S.; Haft, D.H.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Horvath, P.; Moineau, S.; Mojica, F.J.M.; Wolf, Y.I.; Yakunin, A.F.; et al. Evolution and classification of the CRISPR—Cas systems. Nat. Rev. Microbiol. 2011, 9, 467–477.
- DeBoy, R.T.; Mongodin, E.F.; Emerson, J.B.; Nelson, K.E. Chromosome Evolution in the Thermotogales: Large-Scale Inversions and Strain Diversification of CRISPR Sequences. J. Bacteriol. 2006, 188, 2364–2374.
- Jansen, R.; van Embden, J.D.A.; Gaastra, W.; Schouls, L.M. Identification of genes that are associated with DNA repeats in prokaryotes. Mol. Microbiol. 2002, 43, 1565–1575.
- Mojica, F.J.M.; Diez-Villasenor, C.; Soria, E.; Juez, G. Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol. Microbiol. 2000, 36, 244–246.
- Pourcel, C.; Salvignol, G.; Vergnaud, G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 2005, 151, 653–663.
- Hille, F.; Richter, H.; Wong, S.P.; Bratovič, M.; Ressel, S.; Charpentier, E. The Biology of CRISPR-Cas: Backward and Forward. Cell 2018, 172, 1239–1259.
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary classification of CRISPR–Cas systems: A burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020, 18, 67–83.
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR—Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736.
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823.
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821.
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–826.
- Barrangou, R. CRISPR-Cas systems and RNA-guided interference: CRISPR-Cas systems and RNA-guided interference. WIREs RNA 2013, 4, 267–278.
- Bhaya, D.; Davison, M.; Barrangou, R. CRISPR-Cas Systems in Bacteria and Archaea: Versatile Small RNAs for Adaptive Defense and Regulation. Annu. Rev. Genet. 2011, 45, 273–297.
- Yosef, I.; Goren, M.G.; Qimron, U. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012, 40, 5569–5576.
- Nuñez, J.K.; Kranzusch, P.J.; Noeske, J.; Wright, A.V.; Davies, C.W.; Doudna, J.A. Cas1–Cas2 complex formation mediates spacer acquisition during CRISPR–Cas adaptive immunity. Nat. Struct. Mol. Biol. 2014, 21, 528–534.
- Heler, R.; Samai, P.; Modell, J.W.; Weiner, C.; Goldberg, G.W.; Bikard, D.; Marraffini, L.A. Cas9 specifies functional viral targets during CRISPR–Cas adaptation. Nature 2015, 519, 199–202.
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607.
- Thurtle-Schmidt, D.M.; Lo, T.-W. Molecular biology at the cutting edge: A review on CRISPR/CAS9 gene editing for undergraduates: A Review on CRISPR/CAS9 Gene Editing for Undergraduates. Biochem. Mol. Biol. Educ. 2018, 46, 195–205.
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586.
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014, 513, 569–573.
- Jinek, M.; Jiang, F.; Taylor, D.W.; Sternberg, S.H.; Kaya, E.; Ma, E.; Anders, C.; Hauer, M.; Zhou, K.; Lin, S.; et al. Structures of Cas9 Endonucleases Reveal RNA-Mediated Conformational Activation. Science 2014, 343, 1247997.
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal Structure of Cas9 in Complex with Guide RNA and Target DNA. Cell 2014, 156, 935–949.
- Rodriguez-Rodriguez, D.; Ramirez-Solis, R.; Garza-Elizondo, M.; Garza-Rodriguez, M.; Barrera-Saldana, H. Genome editing: A perspective on the application of CRISPR/Cas9 to study human diseases (Review). Int. J. Mol. Med. 2019.
- Biagioni, A.; Chillà, A.; Andreucci, E.; Laurenzana, A.; Margheri, F.; Peppicelli, S.; Del Rosso, M.; Fibbi, G. Type II CRISPR/Cas9 approach in the oncological therapy. J. Exp. Clin. Cancer Res. 2017, 36, 80.
- Jiang, F.; Doudna, J.A. CRISPR–Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529.
- Wyman, C.; Kanaar, R. DNA Double-Strand Break Repair: All’s Well that Ends Well. Annu. Rev. Genet. 2006, 40, 363–383.
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191.
- Martinez-Lage, M.; Puig-Serra, P.; Menendez, P.; Torres-Ruiz, R.; Rodriguez-Perales, S. CRISPR/Cas9 for Cancer Therapy: Hopes and Challenges. Biomedicines 2018, 6, 105.
- Petrucelli, N.; Daly, M.B.; Feldman, G.L. Hereditary breast and ovarian cancer due to mutations in BRCA1 and BRCA2. Genet. Med. 2010, 12, 245–259.
- Huen, M.S.Y.; Sy, S.M.H.; Chen, J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat. Rev. Mol. Cell Biol. 2010, 11, 138–148.
- Brzovic, P.S.; Rajagopal, P.; Hoyt, D.W.; King, M.-C.; Klevit, R.E. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat. Struct. Biol. 2001, 8, 833–837.
- Brzovic, P.S.; Keeffe, J.R.; Nishikawa, H.; Miyamoto, K.; Fox, D.; Fukuda, M.; Ohta, T.; Klevit, R. Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin-ligase complex. Proc. Natl. Acad. Sci. USA 2003, 100, 5646–5651.
- Yamane, K.; Katayama, E.; Tsuruo, T. The BRCT Regions of Tumor Suppressor BRCA1 and of XRCC1 Show DNA End Binding Activity with a Multimerizing Feature. Biochem. Biophys. Res. Commun. 2000, 279, 678–684.
- Liu, J.; Pan, Y.; Ma, B.; Nussinov, R. “Similarity Trap” in Protein-Protein Interactions Could Be Carcinogenic: Simulations of p53 Core Domain Complexed with 53BP1 and BRCA1 BRCT Domains. Structure 2006, 14, 1811–1821.
- Kweon, J.; Jang, A.-H.; Shin, H.R.; See, J.-E.; Lee, W.; Lee, J.W.; Chang, S.; Kim, K.; Kim, Y. A CRISPR-based base-editing screen for the functional assessment of BRCA1 variants. Oncogene 2020, 39, 30–35.
- Corso, G.; Feroce, I.; Intra, M.; Toesca, A.; Magnoni, F.; Sargenti, M.; Naninato, P.; Caldarella, P.; Pagani, G.; Vento, A.; et al. BRCA1/2 germline missense mutations: A systematic review. Eur. J. Cancer Prev. 2018, 27, 279–286.
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2012, 12, 68–78.
- Suwaki, N.; Klare, K.; Tarsounas, M. RAD51 paralogs: Roles in DNA damage signalling, recombinational repair and tumorigenesis. Semin. Cell Dev. Biol. 2011, 22, 898–905.
- Wong, M.W.; Nordfors, C.; Mossman, D.; Pecenpetelovska, G.; Avery-Kiejda, K.A.; Talseth-Palmer, B.; Bowden, N.A.; Scott, R.J. BRIP1, PALB2, and RAD51C mutation analysis reveals their relative importance as genetic susceptibility factors for breast cancer. Breast Cancer Res. Treat. 2011, 127, 853–859.
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous Recombination and Human Health: The Roles of BRCA1, BRCA2, and Associated Proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600.
- Feng, W.; Jasin, M. BRCA2 suppresses replication stress-induced mitotic and G1 abnormalities through homologous recombination. Nat. Commun. 2017, 8, 525.
- Slamon, D.; Clark, G.; Wong, S.; Levin, W.; Ullrich, A.; McGuire, W. Human Breast Cancer: Correlation of Relapse and Survival with Amplification of the HER-2/neu Oncogene. Science 1987, 235, 177–182.
- Wang, H.; Sun, W. CRISPR-mediated targeting of HER2 inhibits cell proliferation through a dominant negative mutation. Cancer Lett. 2017, 385, 137–143.
- Gutierrez, C.; Schiff, R. HER2: Biology, detection, and clinical implications. Arch. Pathol. Lab. Med. 2011, 135, 55–62.
- Yarden, Y. Biology of HER2 and Its Importance in Breast Cancer. Oncology 2001, 61, 1–13.
- Moasser, M.M. The oncogene HER2: Its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 2007, 26, 6469–6487.
- Home—My Cancer Genome. Available online: (accessed on 27 November 2020).
- Welcome—IARC TP53 Database. Available online: (accessed on 27 November 2020).
- Zhu, J.; Sammons, M.A.; Donahue, G.; Dou, Z.; Vedadi, M.; Getlik, M.; Barsyte-Lovejoy, D.; Al-awar, R.; Katona, B.W.; Shilatifard, A.; et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 2015, 525, 206–211.
- Levine, A.J.; Berger, S.L. The interplay between epigenetic changes and the p53 protein in stem cells. Genes Dev. 2017, 31, 1195–1201.
- Metastasis Has Multiple Origins and Occurs Early in Tumorigenesis. Cancer Discov. 2020, 10, 903.
- Belotserkovskaya, R.; Raga Gil, E.; Lawrence, N.; Butler, R.; Clifford, G.; Wilson, M.D.; Jackson, S.P. PALB2 chromatin recruitment restores homologous recombination in BRCA1-deficient cells depleted of 53BP1. Nat. Commun. 2020, 11, 819.
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 Cellular and Clinical Functions by a Nuclear Partner, PALB2. Mol. Cell 2006, 22, 719–729.
- Ripperger, T.; Gadzicki, D.; Meindl, A.; Schlegelberger, B. Breast cancer susceptibility: Current knowledge and implications for genetic counselling. Eur. J. Hum. Genet. 2009, 17, 722–731.
- Wiltshire, T.; Ducy, M.; Foo, T.K.; Hu, C.; Lee, K.Y.; Belur Nagaraj, A.; Rodrigue, A.; Gomes, T.T.; Simard, J.; Monteiro, A.N.A.; et al. Functional characterization of 84 PALB2 variants of uncertain significance. Genet. Med. 2020, 22, 622–632.
- Cidado, J.; Wong, H.Y.; Rosen, D.M.; Cimino-Mathews, A.; Garay, J.P.; Fessler, A.G.; Rasheed, Z.A.; Hicks, J.; Cochran, R.L.; Croessmann, S.; et al. Ki-67 is required for maintenance of cancer stem cells but not cell proliferation. Oncotarget 2016, 7, 6281–6293.
- Castéra, L.; Harter, V.; Muller, E.; Krieger, S.; Goardon, N.; Ricou, A.; Rousselin, A.; Paimparay, G.; Legros, A.; Bruet, O.; et al. Landscape of pathogenic variations in a panel of 34 genes and cancer risk estimation from 5131 HBOC families. Genet. Med. 2018, 20, 1677–1686.
- Chapman, J.R.; Sossick, A.J.; Boulton, S.J.; Jackson, S.P. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J. Cell Sci. 2012, 125, 3529–3534.
- Endl, E.; Gerdes, J. The Ki-67 Protein: Fascinating Forms and an Unknown Function. Exp. Cell Res. 2000, 257, 231–237.
- Cuzick, J.; Dowsett, M.; Pineda, S.; Wale, C.; Salter, J.; Quinn, E.; Zabaglo, L.; Mallon, E.; Green, A.R.; Ellis, I.O.; et al. Prognostic Value of a Combined Estrogen Receptor, Progesterone Receptor, Ki-67, and Human Epidermal Growth Factor Receptor 2 Immunohistochemical Score and Comparison With the Genomic Health Recurrence Score in Early Breast Cancer. JCO 2011, 29, 4273–4278.
- Cuylen, S.; Blaukopf, C.; Politi, A.Z.; Müller-Reichert, T.; Neumann, B.; Poser, I.; Ellenberg, J.; Hyman, A.A.; Gerlich, D.W. Ki-67 acts as a biological surfactant to disperse mitotic chromosomes. Nature 2016, 535, 308–312.
- Sobecki, M.; Mrouj, K.; Camasses, A.; Parisis, N.; Nicolas, E.; Llères, D.; Gerbe, F.; Prieto, S.; Krasinska, L.; David, A.; et al. The cell proliferation antigen Ki-67 organises heterochromatin. eLife 2016, 5, e13722.
- Takagi, M.; Natsume, T.; Kanemaki, M.T.; Imamoto, N. Perichromosomal protein Ki67 supports mitotic chromosome architecture. Genes Cells 2016, 21, 1113–1124.
- Sobecki, M.; Mrouj, K.; Colinge, J.; Gerbe, F.; Jay, P.; Krasinska, L.; Dulic, V.; Fisher, D. Cell-Cycle Regulation Accounts for Variability in Ki-67 Expression Levels. Cancer Res. 2017, 77, 2722–2734.
- Miller, I.; Min, M.; Yang, C.; Tian, C.; Gookin, S.; Carter, D.; Spencer, S.L. Ki67 is a Graded Rather than a Binary Marker of Proliferation versus Quiescence. Cell Rep. 2018, 24, 1105–1112.e5.
- Dexter, D.L.; Kowalski, H.M.; Blazar, B.A.; Fligiel, Z.; Vogel, R.; Heppner, G.H. Heterogeneity of tumor cells from a single mouse mammary tumor. Cancer Res. 1978, 38, 3174–3181.
- Mrouj, K.; Singh, P.; Sobecki, M.; Dubra, G.; Al Ghoul, E.; Aznar, A.; Prieto, S.; Pirot, N.; Bernex, F.; Bordignon, B.; et al. Ki-67 promotes sequential stages of tumourigenesis by enabling cellular plasticity. Cancer Biol. 2019.
- Redig, A.J.; McAllister, S.S. Breast cancer as a systemic disease: A view of metastasis. J. Intern. Med. 2013, 274, 113–126.
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572.
- Ocaña, O.H.; Córcoles, R.; Fabra, Á.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic Colonization Requires the Repression of the Epithelial-Mesenchymal Transition Inducer Prrx1. Cancer Cell 2012, 22, 709–724.
- Maitra, A. Molecular envoys pave the way for pancreatic cancer to invade the liver. Nature 2019, 567, 181–182.
- Massagué, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306.
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784.
- Nieto, M.A. The Ins and Outs of the Epithelial to Mesenchymal Transition in Health and Disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 347–376.
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495.
- Kushwaha, P.P.; Gupta, S.; Singh, A.K.; Kumar, S. Emerging Role of Migration and Invasion Enhancer 1 (MIEN1) in Cancer Progression and Metastasis. Front. Oncol. 2019, 9, 868.
- Kpetemey, M.; Dasgupta, S.; Rajendiran, S.; Das, S.; Gibbs, L.D.; Shetty, P.; Gryczynski, Z.; Vishwanatha, J.K. MIEN1, a novel interactor of Annexin A2, promotes tumor cell migration by enhancing AnxA2 cell surface expression. Mol. Cancer 2015, 14, 156.
- Dasgupta, S.; Wasson, L.M.; Rauniyar, N.; Prokai, L.; Borejdo, J.; Vishwanatha, J.K. Novel gene C17orf37 in 17q12 amplicon promotes migration and invasion of prostate cancer cells. Oncogene 2009, 28, 2860–2872.
- Dasgupta, S.; Cushman, I.; Kpetemey, M.; Casey, P.J.; Vishwanatha, J.K. Prenylated C17orf37 Induces Filopodia Formation to Promote Cell Migration and Metastasis. J. Biol. Chem. 2011, 286, 25935–25946.
- Van Treuren, T.; Vishwanatha, J.K. CRISPR deletion of MIEN1 in breast cancer cells. PLoS ONE 2018, 13, e0204976.
- Shen, F.; Zhang, Y.; Jernigan, D.L.; Feng, X.; Yan, J.; Garcia, F.U.; Meucci, O.; Salvino, J.M.; Fatatis, A. Novel Small-Molecule CX3CR1 Antagonist Impairs Metastatic Seeding and Colonization of Breast Cancer Cells. Mol. Cancer Res. 2016, 14, 518–527.
- Bazan, J.F.; Bacon, K.B.; Hardiman, G.; Wang, W.; Soo, K.; Rossi, D.; Greaves, D.R.; Zlotnik, A.; Schall, T.J. A new class of membrane-bound chemokine with a CX3C motif. Nature 1997, 385, 640–644.
- Onitilo, A.A.; Engel, J.M.; Greenlee, R.T.; Mukesh, B.N. Breast Cancer Subtypes Based on ER/PR and Her2 Expression: Comparison of Clinicopathologic Features and Survival. Clin. Med. Res. 2009, 7, 4–13.
- Gruber, I.; Fehm, T.; Taran, F.A.; Wallwiener, M.; Hahn, M.; Wallwiener, D.; Krawzyck, N.; Hoffmann, J.; Hartkopf, A.D. Disseminated tumor cells as a monitoring tool for adjuvant therapy in patients with primary breast cancer. Breast Cancer Res. Treat. 2014, 144, 353–360.
- Cook, D.N.; Chen, S.-C.; Sullivan, L.M.; Manfra, D.J.; Wiekowski, M.T.; Prosser, D.M.; Vassileva, G.; Lira, S.A. Generation and Analysis of Mice Lacking the Chemokine Fractalkine. Mol. Cell. Biol. 2001, 21, 3159–3165.
- Jamieson-Gladney, W.L.; Zhang, Y.; Fong, A.M.; Meucci, O.; Fatatis, A. The chemokine receptor CX3CR1 is directly involved in the arrest of breast cancer cells to the skeleton. Breast Cancer Res. 2011, 13, R91.
- Xie, K. Interleukin-8 and human cancer biology. Cytokine Growth Factor Rev. 2001, 12, 375–391.
- Long, X.; Ye, Y.; Zhang, L.; Liu, P.; Yu, W.; Wei, F.; Ren, X.; Yu, J. IL-8, a novel messenger to cross-link inflammation and tumor EMT via autocrine and paracrine pathways (Review). Int. J. Oncol. 2016, 48, 5–12.
- Stillie, R.; Farooq, S.M.; Gordon, J.R.; Stadnyk, A.W. The functional significance behind expressing two IL-8 receptor types on PMN. J. Leukoc. Biol. 2009, 86, 529–543.
- Wuyts, A.; Van Osselaer, N.; Haelens, A.; Samson, I.; Herdewijn, P.; Ben-Baruch, A.; Oppenheim, J.J.; Proost, P.; Van Damme, J. Characterization of Synthetic Human Granulocyte Chemotactic Protein 2: Usage of Chemokine Receptors CXCR1 and CXCR2 and in Vivo Inflammatory Properties. Biochemistry 1997, 36, 2716–2723.
- Saintigny, P.; Massarelli, E.; Lin, S.; Ahn, Y.-H.; Chen, Y.; Goswami, S.; Erez, B.; O’Reilly, M.S.; Liu, D.; Lee, J.J.; et al. CXCR2 Expression in Tumor Cells Is a Poor Prognostic Factor and Promotes Invasion and Metastasis in Lung Adenocarcinoma. Cancer Res. 2013, 73, 571–582.
- Jin, K.; Pandey, N.B.; Popel, A.S. Crosstalk between stromal components and tumor cells of TNBC via secreted factors enhances tumor growth and metastasis. Oncotarget 2017, 8, 60210–60222.
- Sun, X.; Cheng, G.; Hao, M.; Zheng, J.; Zhou, X.; Zhang, J.; Taichman, R.S.; Pienta, K.J.; Wang, J. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010, 29, 709–722.
- Yang, M.; Zeng, C.; Li, P.; Qian, L.; Ding, B.; Huang, L.; Li, G.; Jiang, H.; Gong, N.; Wu, W. Impact of CXCR4 and CXCR7 knockout by CRISPR/Cas9 on the function of triple-negative breast cancer cells. OncoTargets Ther. 2019, 12, 3849–3858.
- Ganju, R.K.; Brubaker, S.A.; Meyer, J.; Dutt, P.; Yang, Y.; Qin, S.; Newman, W.; Groopman, J.E. The α-Chemokine, Stromal Cell-derived Factor-1α, Binds to the Transmembrane G-protein-coupled CXCR-4 Receptor and Activates Multiple Signal Transduction Pathways. J. Biol. Chem. 1998, 273, 23169–23175.
- Miao, Z.; Luker, K.E.; Summers, B.C.; Berahovich, R.; Bhojani, M.S.; Rehemtulla, A.; Kleer, C.G.; Essner, J.J.; Nasevicius, A.; Luker, G.D.; et al. CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc. Natl. Acad. Sci. USA 2007, 104, 15735–15740.
- Wang, J.; Shiozawa, Y.; Wang, J.; Wang, Y.; Jung, Y.; Pienta, K.J.; Mehra, R.; Loberg, R.; Taichman, R.S. The Role of CXCR7/RDC1 as a Chemokine Receptor for CXCL12/SDF-1 in Prostate Cancer. J. Biol. Chem. 2008, 283, 4283–4294.
- Zheng, K.; Li, H.-Y.; Su, X.-L.; Wang, X.-Y.; Tian, T.; Li, F.; Ren, G.-S. Chemokine receptor CXCR7 regulates the invasion, angiogenesis and tumor growth of human hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. 2010, 29, 31.
- Burns, J.M.; Summers, B.C.; Wang, Y.; Melikian, A.; Berahovich, R.; Miao, Z.; Penfold, M.E.T.; Sunshine, M.J.; Littman, D.R.; Kuo, C.J.; et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J. Exp. Med. 2006, 203, 2201–2213.
- Wu, W.; Qian, L.; Dai, J.; Ding, B.; Chen, X. Expression of chemokine CXCL12 and its receptor (CXCR4 and CXCR7) in different molecular subtypes of human breast carcinoma and the clinical significance. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2017, 42, 147–153.
- Johnson, G.L. Mitogen-Activated Protein Kinase Pathways Mediated by ERK, JNK, and p38 Protein Kinases. Science 2002, 298, 1911–1912.
- Rattanasinchai, C.; Gallo, K. MLK3 Signaling in Cancer Invasion. Cancers 2016, 8, 51.
- Cronan, M.R.; Nakamura, K.; Johnson, N.L.; Granger, D.A.; Cuevas, B.D.; Wang, J.-G.; Mackman, N.; Scott, J.E.; Dohlman, H.G.; Johnson, G.L. Defining MAP3 kinases required for MDA-MB-231 cell tumor growth and metastasis. Oncogene 2012, 31, 3889–3900.
- Jiang, X.; Xie, H.; Dou, Y.; Yuan, J.; Zeng, D.; Xiao, S. Expression and function of FRA1 protein in tumors. Mol. Biol. Rep. 2020, 47, 737–752.
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67.
- Rattanasinchai, C.; Llewellyn, B.J.; Conrad, S.E.; Gallo, K.A. MLK3 regulates FRA-1 and MMPs to drive invasion and transendothelial migration in triple-negative breast cancer cells. Oncogenesis 2017, 6, e345.
- Kohli, S.; Kohli, V. Role of RANKL-RANK/osteoprotegerin molecular complex in bone remodeling and its immunopathologic implications. Indian J. Endocr. Metab. 2011, 15, 175.
- Cody, J.J.; Rivera, A.A.; Lyons, G.R.; Yang, S.W.; Wang, M.; Sarver, D.B.; Wang, D.; Selander, K.S.; Kuo, H.-C.; Meleth, S.; et al. Arming a replicating adenovirus with osteoprotegerin reduces the tumor burden in a murine model of osteolytic bone metastases of breast cancer. Cancer Gene 2010, 17, 893–905.
- Sheridan, J.P. Control of TRAIL-Induced Apoptosis by a Family of Signaling and Decoy Receptors. Science 1997, 277, 818–821.
- Geerts, D.; Chopra, C.; Connelly, L. Osteoprotegerin: Relationship to Breast Cancer Risk and Prognosis. Front. Oncol. 2020, 10, 462.
- Zhong, Z.; Niu, P.; Wang, M.; Huang, G.; Xu, S.; Sun, Y.; Xu, X.; Hou, Y.; Sun, X.; Yan, Y.; et al. Targeted disruption of sp7 and myostatin with CRISPR-Cas9 results in severe bone defects and more muscular cells in common carp. Sci. Rep. 2016, 6, 22953.
- Kichev, A.; Eede, P.; Gressens, P.; Thornton, C.; Hagberg, H. Implicating Receptor Activator of NF-κB (RANK)/RANK Ligand Signalling in Microglial Responses to Toll-Like Receptor Stimuli. Dev. Neurosci. 2017, 39, 192–206.
- Goswami, S.; Sharma-Walia, N. Crosstalk between osteoprotegerin (OPG), fatty acid synthase (FASN) and, cycloxygenase-2 (COX-2) in breast cancer: Implications in carcinogenesis. Oncotarget 2016, 7, 58953–58974.
- Song, L.; Luo, Z.-Q. Post-translational regulation of ubiquitin signaling. J. Cell Biol. 2019, 218, 1776–1786.
- Shearer, R.F.; Iconomou, M.; Watts, C.K.W.; Saunders, D.N. Functional Roles of the E3 Ubiquitin Ligase UBR5 in Cancer. Mol. Cancer Res. 2015, 13, 1523–1532.
- Liao, L.; Song, M.; Li, X.; Tang, L.; Zhang, T.; Zhang, L.; Pan, Y.; Chouchane, L.; Ma, X. E3 Ubiquitin Ligase UBR5 Drives the Growth and Metastasis of Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 2090–2101.
- Mavaddat, N.; Barrowdale, D.; Andrulis, I.L.; Domchek, S.M.; Eccles, D.; Nevanlinna, H.; Ramus, S.J.; Spurdle, A.; Robson, M.; Sherman, M.; et al. Pathology of Breast and Ovarian Cancers among BRCA1 and BRCA2 Mutation Carriers: Results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). Cancer Epidemiol. Biomark. Prev. 2012, 21, 134–147.
- Wilson, J.R.F.; Bateman, A.C.; Hanson, H.; An, Q.; Evans, G.; Rahman, N.; Jones, J.L.; Eccles, D.M. A novel HER2-positive breast cancer phenotype arising from germline TP53 mutations. J. Med. Genet. 2010, 47, 771–774.
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695.
- Ahmed, A.; Ashraf, D.; Bahaa, A.; El-Tayebi, H.; Adwan, H. Impact of CDK4 knock out using CRISPR/Cas9 gene editing technology on breast cancer progression. Eur. J. Cancer 2020, 138, S70–S71.
- Xu, K.; Chen, G.; Li, X.; Wu, X.; Chang, Z.; Xu, J.; Zhu, Y.; Yin, P.; Liang, X.; Dong, L. MFN2 suppresses cancer progression through inhibition of mTORC2/Akt signaling. Sci. Rep. 2017, 7, 41718.
- Mendes de Almeida, R.; Bandarra, S.; Clara Ribeiro, A.; Mascarenhas, P.; Bekman, E.; Barahona, I. Inactivation of APOBEC3G gene in breast cancer cells using the CRISPR/Cas9 system. Ann. Med. 2019, 51, 40.
- Heidary Arash, E.; Shiban, A.; Song, S.; Attisano, L. MARK4 inhibits Hippo signaling to promote proliferation and migration of breast cancer cells. EMBO Rep. 2017, 18, 420–436.
- Álvarez-Fernández, M.; Sanz-Flores, M.; Sanz-Castillo, B.; Salazar-Roa, M.; Partida, D.; Zapatero-Solana, E.; Ali, H.R.; Manchado, E.; Lowe, S.; VanArsdale, T.; et al. Therapeutic relevance of the PP2A-B55 inhibitory kinase MASTL/Greatwall in breast cancer. Cell Death Differ. 2017.
- Lin, A.; Giuliano, C.J.; Sayles, N.M.; Sheltzer, J.M. CRISPR/Cas9 mutagenesis invalidates a putative cancer dependency targeted in on-going clinical trials. eLife 2017, 6, e24179.
- Tian, J.; Wang, V.; Wang, N.; Khadang, B.; Boudreault, J.; Bakdounes, K.; Ali, S.; Lebrun, J.-J. Identification of MFGE8 and KLK5/7 as mediators of breast tumorigenesis and resistance to COX-2 inhibition. Breast Cancer Res. 2021, 23, 23.


