A major breakthrough in the field of genomics is the development of the CRISPR/Cas9 technology that has revolutionized gene editing in the 21st century. CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) was first identified in Escherichia Coli in 1987 as a group of repeated fragments comprised of 29 nucleotides that are separated by fragments of 32 nucleotides of unique varied sequence [
1]. This was shown to play a role in multiple cellular processes including thermal adaptation [
15], DNA repair [
4] and chromosomal rearrangements [
16]. In addition, a comparable 24 to 40 nucleotide short palindromic repeat sequence interspaced by a 20 to 58 varied nucleotide sequence was later identified in multiple species of bacteria and archaea, such as in Streptococcus pyogenes (S.pyogenes), Mycobacterium tuberculosis and Haloferax Mediterranean [
17,
18]. In 2005, researchers elucidated the homology between the short spacer fragments found on the CRISPR locus and the DNA of prokaryotic invading pathogens. Research over the years showed that CRISPR evolved with time as an adaptive immune system, protecting bacteria and archaea from foreign DNA invaders such as viruses and plasmids [
19].
The CRISPR/Cas systems are grouped into 2 classes, 6 types, and 33 subtypes indicated by the involvement of the different Cas proteins within the CRISPR framework that either target DNA, RNA, or both [
8,
20]. The classification is summarized in [
21].
Table 1. Classification of the CRISPR/Cas Systems.
| CRIPSR/Cas Systems |
| Class |
1 |
2 |
| Protein type |
Multiplex |
Single |
| Type |
I |
III |
IV |
II |
V |
VI |
Corresponding
Cas protein |
Cas 3 |
Cas 10 |
Cas 8 |
Cas 9 |
Cas 12a, Cas 12c, Cas 13a |
Cas 13b,
Cas 13c |
In 2013, scientists proposed the development of a targeted genome editing tool using the CRISPR/Cas9 technology found in S. pyogenes [
10]. Specifically, the class 2 type II subgroup found in this species is most extensively employed for genome editing due to its simplicity necessitating merely a single Cas protein, the endonuclease protein Cas9, along with 2 RNA components, CRISPR RNA (crRNA) and trans-activating CRISPR RNA (tracrRNA). The CRISPR/Cas9 system in S. pyogenes (SpCas9) was further simplified to constitute two components, the Cas9 protein and a single guide RNA (sgRNA) through the hybridization of crRNA and tracrRNA, enabling the manipulation of the eukaryotic genome [
10,
22,
23]. Correspondingly, the immunity provided by the CRISPR/Cas9 system can be characterized into three phases: (1) integration of phage or plasmid DNA into the CRISPR array, (2) CRISPR locus transcription to form pre-crRNA and maturation into crRNA and formation of tracrRNA, and (3) DNA manipulation [
24].
The initial phase of CRISPR/Cas9 activity is the integration of short sequences of phage or plasmid DNA, termed protospacer, into the host genome which serves as a cellular memory of past infections. This enables prokaryotes to distinguish subsequent infections by these invaders as foreign, leading to silencing of the alien DNA. The acquired foreign DNA constitutes the varied region, called spacer, found on the CRISPR loci [
25]. CRISPR spacer acquisition is mediated by two core proteins, Cas1 and Cas2, which are the only proteins virtually found in almost all the identified CRISPR/Cas systems [
26]. A stable complex is formed between these two proteins to initiate the adaptation process; Cas1 possesses endonuclease activity that is necessary for spacer integration while Cas2 seems to play out a non-enzymatic role [
27]. It has also been suggested that Cas9 plays a direct role in protospacer acquisition by recruiting Cas1 and Cas2 to potential targets [
28].
The second phase begins with the transcription of the CRISPR locus to generate pre-crRNA, a long RNA molecule that contains sequences complementary to those of the spacers and repeats. The tracrRNA is the second RNA molecule needed and is essential for pre-crRNA maturation, it is transcribed from a genomic locus located upstream of the CRISPR locus [
29]. The tracrRNA contains a segment that is homologous to the cognate sequence of the CRISPR locus; therefore, is able to bind to the 3′ end of the pre-crRNA forming a double-stranded RNA molecule [
30]. Subsequently, the pre-crRNA:tracrRNA double-stranded RNA is cleaved by recruited cellular ribonuclease III (RNase III) responsible for the recognition and cleavage of double-stranded RNA molecules [
29]. A second cleavage takes place whereby the 5′ end of the RNA sequence is cut, yielding a mature crRNA:tracrRNA (gRNA) complex ready to associate with a Cas protein, with each individual crRNA fragment containing a unique spacer sequence that is around 20 nucleotides in length [
30,
31]. The resulting gRNA complex binds to the Cas9 protein, creating a Cas9:gRNA effector complex capable of DNA interference to complete the CRISPR mediated immunity. The Cas9 protein is a dual RNA-guided endonuclease enzyme having a bi-lobed structure, the α-helical recognition (REC) lobe and the nuclease lobe, with the RNA complex situated in between. The latter is comprised of two nuclease domains, an HNH domain responsible for cleaving the complementary DNA strand to crRNA and a RuvC-like domain which cleaves the non-complementary DNA strand. On the other hand, the REC lobe contains an arginine-rich bridge that is essential for RNA interaction and joining the two lobes together [
32,
33,
34]. Once Cas9 is activated by binding to the gRNA complex, it scavenges for any invading nucleic acid sequences that show complementarity to the crRNA. Therefore, CRISPR/Cas9 initiates a double-stranded cleavage at a specified DNA sequence site following base pairing of crRNA to the target site [
35] ().
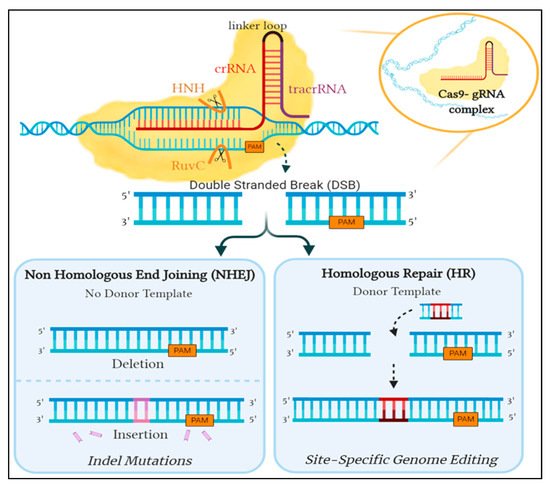
Figure 1. An overview of the repair mechanism associated with induced Cas9 double-stranded DNA break. Cleavage is induced by the binding of Cas9- gRNA complex to its complementary sequence on foreign DNA. In eukaryotes, this is amended by either of two mechanisms: the error-prone Non-Homologous End Joining (NHEJ) or Homologous Repair (HR), which is utilized for genome editing by providing a donor template. Created with Biorender.
However, the prospective target sequence is only valid if a short sequence known as Protospacer Adjacent Motif (PAM) is present directly after the binding location of crRNA. The presence of PAM is the underlying factor that determines preference between self and non-self DNA. Although the CRISPR array contains spacers that are identical to foreign DNA, the CRISPR genome is not affected by its own mechanism as the spacers do not lie immediately next to a PAM sequence [
36]. In addition, the PAM sequence identified by Cas9 varies between microorganisms; SpCas9 specifically recognizes 5′-NGG-3′ [
35], resulting in a blunt-end double-strand break occurring upstream by three base pairs in the PAM sequence [
30]. The guanine dinucleotide [
32] of PAM found on the non-complementary strand aids in its recognition by interacting with two crucial arginine residues. Further interactions form a bend in the target DNA assisting in the unwinding of the helical structure which propagates cutting of the intruder DNA [
37]. This disruption in the invading pathogen is deemed to be detrimental to its existence and is ultimately what provides protection for the prokaryote.
This guided interference into the DNA sequence inspired researchers to exploit the system with hopes of achieving precise genome editing. Unfortunately, the CRISPR/Cas9 system in prokaryotes utilizes components not inherently present in eukaryotes, prompting the need to optimize the S. pyogenes’ CRISPR/Cas9 system. The modifiable crRNA is merged with the tracrRNa to form sgRNA, which works similarly to the gRNA complex as it guides Cas9 to the target sequence site and triggers the cleavage of both DNA strands. The double-strand DNA breaks induced by Cas9 can then be amended by one of two DNA repair pathways: either the non-homologous end joining (NHEJ) or the homology-directed repair (HDR) [
38]. NHEJ ligates the broken ends together, however, this pathway is error-prone and could lead to insertion/deletion (indel) mutations resulting in an ineffective gene. In contrast, HDR uses a neighboring homologous sequence as a template to mend the break. This method can be exploited to potentially introduce targeted edits at a precise location into the DNA sequence by providing a donor template attached to sgRNA for repair [
30] ().
As the machinery behind the whole CRISPR/Cas9 system relies on the complementarity between crRNA and the target sequence, in addition to the presence of PAM [
35], specificity is crucial when generating a sgRNA. In case the desired target is inaccurately outlined, Cas9 can bind and cause an off-target cleave of the sequence, leading to unintended mutations that could be consequential. However, as long as the target sequence is identified, several CRISPR software tools are available to facilitate the design of an optimal sgRNA to achieve precise cleavage with minimal off-target effects [
39]. Owing to the versatility in sequencing sgRNAs, attachment of the Cas9:gRNA complex to various sites is plausible. This merits the CRISPR/Cas9 system to reconstruct a multitude of loci concurrently [
40].