 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Silvia Di Agostino | + 4127 word(s) | 4127 | 2021-06-04 10:55:40 | | | |
| 2 | Vivi Li | Meta information modification | 4127 | 2021-06-04 11:29:39 | | |
Video Upload Options
Long non-coding RNAs (lncRNAs), circular RNAs (circRNAs), micro RNAs (miRNAs), and extracellular RNAs (exRNAs) are new groups of RNAs with regulation activities that have low or no protein-coding ability. Emerging evidence suggests that deregulated expression of these non-coding RNAs is associated with the induction and progression of diverse tumors throughout epigenetic, transcriptional, and post-transcriptional modifications. A consistent number of non-coding RNAs (ncRNAs) has been shown to be regulated by p53, the most important tumor suppressor of the cells frequently mutated in human cancer. It has been shown that some mutant p53 proteins are associated with the loss of tumor suppressor activity and the acquisition of new oncogenic functions named gain-of-function activities.
1. Introduction
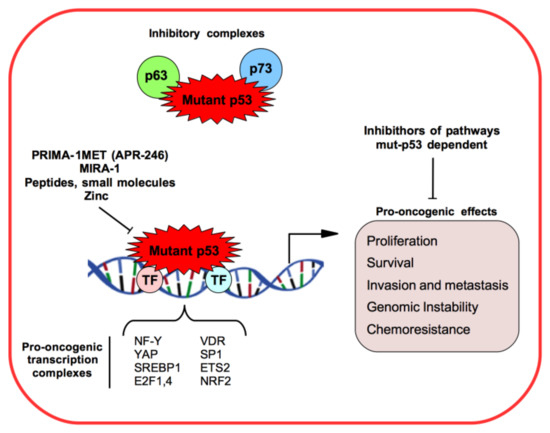
1.1. Mutant p53 Gain-of-Function
1.2. Non-Coding RNAs in Cancer
2. Mutant p53 and miRNAs
2.1. miRNAs Induced by Mutant p53
2.2. miRNAs Inhibited by Mutant p53
3. Mutant p53 and Long Non-Coding RNAs
References
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431.
- Bieging, K.T.; Mello, S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370.
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615.
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339.
- Walerych, D.; Lisek, K.; Del Sal, G. Mutant p53: One, No One, and One Hundred Thousand. Front. Oncol. 2015, 5, 466.
- Guha, T.; Malkin, D. Inherited TP53 Mutations and the Li–Fraumeni Syndrome. Cold Spring Harb. Perspect. Med. 2017, 7, a026187.
- Goh, A.M.; Coffill, C.; Lane, D.P. The role of mutant p53 in human cancer. J. Pathol. 2010, 223, 116–126.
- Brosh, R.; Rotter, V. When mutants gain new powers: news from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713.
- Strano, S.; Dell’Orso, S.; Di Agostino, S.; Fontemaggi, G.; Sacchi, A.; Blandino, G. Mutant p53: an oncogenic transcription factor. Oncogene 2007, 26, 2212–2219.
- Bargonetti, J.; Prives, C. Gain-of-function mutant p53: history and speculation. J. Mol. Cell Boil. 2019, 11, 605–609.
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.-K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 2019, 28, 1370–1384.
- Freed-Pastor, W.; Mizuno, H.; Zhao, X.; Langerød, A.; Moon, S.-H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 Disrupts Mammary Tissue Architecture via the Mevalonate Pathway. Cell 2012, 148, 244–258.
- Zhang, C.; Liu, J.; Liang, Y.; Wu, R.; Zhao, Y.; Hong, X.; Lin, M.; Yu, H.; Liu, L.; Levine, A.J.; et al. Tumour-associated mutant p53 drives the Warburg effect. Nat. Commun. 2013, 4, 2935.
- Di Agostino, S.; Fontemaggi, G.; Strano, S.; Blandino, G.; Orazi, G.D. Targeting mutant p53 in cancer: the latest insights. J. Exp. Clin. Cancer Res. 2019, 38, 290.
- Blandino, G.; Valenti, F.; Sacconi, A.; Di Agostino, S. Wild type- and mutant p53 proteins in mitochondrial dysfunction: Emerging insights in cancer disease. Semin. Cell Dev. Boil. 2020, 98, 105–117.
- Di Agostino, S.; Strano, S.; Emiliozzi, V.; Zerbini, V.; Mottolese, M.; Sacchi, A.; Blandino, G.; Piaggio, G. Gain of function of mutant p53: The mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 2006, 10, 191–202.
- Stambolsky, P.; Tabach, Y.; Fontemaggi, G.; Weisz, L.; Maor-Aloni, R.; Siegfried, Z.; Shiff, I.; Kogan, I.; Shay, M.; Kalo, E.; et al. Modulation of the Vitamin D3 Response by Cancer-Associated Mutant p53. Cancer Cell 2010, 17, 523.
- Di Agostino, S.; Sorrentino, G.; Ingallina, E.; Valenti, F.; Ferraiuolo, M.; Bicciato, S.; Piazza, S.; Strano, S.; Del Sal, G.; Blandino, G. YAP enhances the pro-proliferative transcriptional activity of mutant p53 proteins. EMBO Rep. 2015, 17, 188–201.
- Valenti, F.; Ganci, F.; Fontemaggi, G.; Sacconi, A.; Strano, S.; Blandino, G.; Di Agostino, S. Gain of function mutant p53 proteins cooperate with E2F4 to transcriptionally downregulate RAD17 and BRCA1 gene expression. Oncotarget 2015, 6, 5547–5566.
- Ferraiuolo, M.; Di Agostino, S.; Blandino, G.; Strano, S. Oncogenic Intra-p53 Family Member Interactions in Human Cancers. Front. Oncol. 2016, 6, 305.
- Brown, C.J.; Cheok, C.F.; Verma, C.S.; Lane, D.P. Reactivation of p53: from peptides to small molecules. Trends Pharmacol. Sci. 2011, 32, 53–62.
- Muller, P.A.J.; Vousden, K.H. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317.
- Blandino, G.; Di Agostino, S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J. Exp. Clin. Cancer Res. 2018, 37, 30.
- Garufi, A.; Pucci, D.; D’Orazi, V.; Cirone, M.; Bossi, G.; Avantaggiati, M.L.; Orazi, G.D. Degradation of mutant p53H175 protein by Zn(II) through autophagy. Cell Death Dis. 2014, 5, e1271.
- Kogan, S.; Carpizo, D.R. Zinc Metallochaperones as Mutant p53 Reactivators: A New Paradigm in Cancer Therapeutics. Cancers 2018, 10, 166.
- Cao, X.; Hou, J.; An, Q.; Assaraf, Y.G.; Wang, X. Towards the overcoming of anticancer drug resistance mediated by p53 mutations. Drug Resist. Updat. 2020, 49, 100671.
- Bykov, V.J.N.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.M.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288.
- Wassman, C.D.; Baronio, R.; Özlem, D.; Wallentine, B.D.; Chen, C.-K.; Hall, L.V.; Salehi, F.; Lin, D.W.; Chung, B.P.; Hatfield, W.; et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 2013, 4, 1407.
- Deveson, I.; Hardwick, S.; Mercer, T.R.; Mattick, J. The Dimensions, Dynamics, and Relevance of the Mammalian Noncoding Transcriptome. Trends Genet. 2017, 33, 464–478.
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.M.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108.
- Castro-Giner, F.; Gkountela, S.; Donato, C.; Alborelli, I.; Quagliata, L.; Ng, C.K.Y.; Piscuoglio, S.; Aceto, N. Cancer Diagnosis Using a Liquid Biopsy: Challenges and Expectations. Diagnostics 2018, 8, 31.
- Kai, K.; Dittmar, R.; Sen, S. Secretory microRNAs as biomarkers of cancer. Semin. Cell Dev. Boil. 2018, 78, 22–36.
- Levin, A.A. Treating Disease at the RNA Level with Oligonucleotides. N. Engl. J. Med. 2019, 380, 57–70.
- Wu, S.; Lopez-Berestein, G.; Calin, G.A.; Sood, A.K. RNAi Therapies: Drugging the Undruggable. Sci. Transl. Med. 2014, 6, 240ps7.
- Adams, B.D.; Kasinski, A.L.; Slack, F.J. Aberrant regulation and function of microRNAs in cancer. Curr. Biol. 2014, 24, R762–R776.
- Wurster, C.D.; Winter, B.; Wollinsky, K.; Ludolph, A.C.; Uzelac, Z.; Witzel, S.; Schocke, M.; Schneider, R.; Kocak, T. Intrathecal administration of nusinersen in adolescent and adult SMA type 2 and 3 patients. J. Neurol. 2018, 266, 183–194.
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-coding RNAs in Oncology. Cell 2019, 179, 1033–1055.
- Roadmap Epigenomics Consortium; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330.
- Tan, L.; Mai, D.; Zhang, B.; Jiang, X.-B.; Zhang, J.; Bai, R.; Ye, Y.; Li, M.; Pan, L.; Su, J.; et al. PIWI-interacting RNA-36712 restrains breast cancer progression and chemoresistance by interaction with SEPW1 pseudogene SEPW1P RNA. Mol. Cancer 2019, 18, 9.
- Bartel, B. Metazoan MicroRNAs. Cell 2018, 173, 20–51.
- Goeman, F.; Strano, S.; Blandino, G. MicroRNAs as Key Effectors in the p53 Network. Biol. Endoplasmic Reticul. 2017, 333, 51–90.
- Hammond, S.M. An overview of microRNAs. Adv. Drug Deliv. Rev. 2015, 87, 3–14.
- Jones, M.F.; Lal, A. MicroRNAs, wild-type and mutant p53: More questions than answers. RNA Boil. 2012, 9, 781–791.
- Ganci, F.; Sacconi, A.; Ben-Moshe, N.B.; Manciocco, V.; Sperduti, I.; Strigari, L.; Covello, R.; Benevolo, M.; Pescarmona, E.; Domany, E.; et al. Expression of TP53 mutation-associated microRNAs predicts clinical outcome in head and neck squamous cell carcinoma patients. Ann. Oncol. 2013, 24, 3082–3088.
- Landi, M.T.; Zhao, Y.; Rotunno, M.; Koshiol, J.; Liu, H.; Bergen, A.; Rubagotti, M.; Goldstein, A.M.; Linnoila, I.; Marincola, F.M.; et al. MicroRNA expression differentiates histology and predicts survival of lung cancer. Clin. Cancer Res. 2010, 16, 430–441.
- Grespi, F.; Landré, V.; Molchadsky, A.; Di Daniele, N.; Marsella, L.T.; Melino, G.; Rotter, V. Differential regulated microRNA by wild type and mutant p53 in induced pluripotent stem cells. Cell Death Dis. 2016, 7, e2567.
- Neilsen, P.; Noll, J.; Mattiske, S.; Bracken, C.P.; Gregory, P.; Schulz, R.B.; Lim, S.P.; Kumar, R.; Suetani, R.J.; Goodall, G.J.; et al. Mutant p53 drives invasion in breast tumors through up-regulation of miR-155. Oncogene 2012, 32, 2992–3000.
- Donzelli, S.; Fontemaggi, G.; Fazi, F.; Di Agostino, S.; Padula, F.; Biagioni, F.; Muti, P.; Strano, S.; Blandino, G. MicroRNA-128-2 targets the transcriptional repressor E2F5 enhancing mutant p53 gain of function. Cell Death Differ. 2011, 19, 1038–1048.
- Valenti, F.; Sacconi, A.; Ganci, F.; Grasso, G.; Strano, S.; Blandino, G.; Di Agostino, S. The miR-205-5p/BRCA1/RAD17 Axis Promotes Genomic Instability in Head and Neck Squamous Cell Carcinomas. Cancers 2019, 11, E1347.
- Cooks, T.; Pateras, I.S.; Jenkins, L.M.; Patel, K.M.; Robles, A.; Morris, J.; Forshew, T.; Appella, E.; Gorgoulis, V.G.; Harris, C.C. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 2018, 9, 771.
- Poeta, M.L.; Manola, J.; Goldwasser, M.A.; Forastiere, A.; Benoit, N.; Califano, J.A.; Ridge, J.A.; Goodwin, J.; Kenady, D.; Saunders, J.; et al. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2007, 357, 2552–2561.
- Skinner, H.D.; Sandulache, V.C.; Ow, T.J.; Meyn, R.E.; Yordy, J.S.; Beadle, B.M.; Fitzgerald, A.L.; Giri, U.; Ang, K.K.; Myers, J.N. TP53 disruptive mutations lead to head and neck cancer treatment failure through inhibition of radiation-induced senescence. Clin. Cancer Res. 2011, 18, 290–300.
- Tucci, P.; Agostini, M.; Grespi, F.; Markert, E.K.; Terrinoni, A.; Vousden, K.H.; Muller, P.A.J.; Dötsch, V.; Kehrloesser, S.; Sayan, B.S.; et al. Loss of p63 and its microRNA-205 target results in enhanced cell migration and metastasis in prostate cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 15312–15317.
- Zhang, Y.; Hu, Y.; Fang, J.-Y.; Xu, J. Gain-of-function miRNA signature by mutant p53 associates with poor cancer outcome. Oncotarget 2016, 7, 11056–11066.
- Qian, B.-Z.; Condeelis, J.S. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51.
- Corney, D.C.; Hwang, C.-I.; Matoso, A.; Vogt, M.; Flesken-Nikitin, A.; Godwin, A.K.; Kamat, A.A.; Sood, A.K.; Ellenson, L.H.; Hermeking, H.; et al. Frequent downregulation of miR-34 family in human ovarian cancers. Clin. Cancer Res. 2010, 16, 1119–1128.
- Kasinski, A.L.; Slack, F.J. miRNA-34 prevents cancer initiation and progression in a therapeutically resistant K-ras and p53-induced mouse model of lung adenocarcinoma. Cancer Res. 2012, 72, 5576–5587.
- Wiggins, J.F.; Ruffino, L.; Kelnar, K.; Omotola, M.; Patrawala, L.; Brown, D.; Bader, A.G. Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res. 2010, 70, 5923–5930.
- Masciarelli, S.; Fontemaggi, G.; Di Agostino, S.; Donzelli, S.; Carcarino, E.; Strano, S.; Blandino, G. Gain-of-function mutant p53 downregulates miR-223 contributing to chemoresistance of cultured tumor cells. Oncogene 2013, 33, 1601–1608.
- Luo, P.; Wang, Q.; Ye, Y.; Zhang, J.; Lu, D.; Cheng, L.; Zhou, H.; Xie, M.; Wang, B. MiR-223-3p functions as a tumor suppressor in lung squamous cell carcinoma by miR-223-3p-mutant p53 regulatory feedback loop. J. Exp. Clin. Cancer Res. 2019, 38, 74.
- Subramanian, M.; Francis, P.; Bilke, S.; Li, X.L.; Hara, T.; Lu, X.; Jones, M.F.; Walker, R.L.; Zhu, Y.; Pineda, M.; et al. A mutant p53/let-7i-axis-regulated gene network drives cell migration, invasion and metastasis. Oncogene 2015, 34, 1094–1104.
- Zhang, L.; Liao, Y.; Tang, L. MicroRNA-34 family: a potential tumor suppressor and therapeutic candidate in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 53.
- Wahid, F.; Shehzad, A.; Khan, T.; Kim, Y.Y. MicroRNAs: Synthesis, mechanism, function, and recent clinical trials. Biochim. et Biophys. Acta BBA Bioenerg. 2010, 1803, 1231–1243.
- Seo, H.A.; Moeng, S.; Sim, S.; Kuh, H.; Choi, S.Y.; Park, J.K. MicroRNA-Based Combinatorial Cancer Therapy: Effects of MicroRNAs on the Efficacy of Anti-Cancer Therapies. Cells 2019, 9, 29.
- Lin, S.; Yu, L.; Song, X.; Bi, J.; Jiang, L.; Wang, Y.; He, M.; Xiao, Q.; Sun, M.; Olopade, O.I.; et al. Intrinsic adriamycin resistance in p53-mutated breast cancer is related to the miR-30c/FANCF/REV1-mediated DNA damage response. Cell Death Dis. 2019, 10, 666.
- Frixa, T.; Sacconi, A.; Cioce, M.; Roscilli, G.; Ferrara, F.F.; Aurisicchio, L.; Pulito, C.; Telera, S.; Carosi, M.A.; Muti, P.; et al. MicroRNA-128-3p-mediated depletion of Drosha promotes lung cancer cell migration. Carcinogenesis 2017, 39, 293–304.
- Muller, P.A.J.; Trinidad, A.G.; Caswell, P.T.; Norman, J.; Vousden, K.H. Mutant p53 regulates Dicer through p63-dependent and -independent mechanisms to promote an invasive phenotype. J. Boil. Chem. 2013, 289, 122–132.
- Garibaldi, F.; Falcone, E.; Trisciuoglio, D.; Colombo, T.; Lisek, K.; Walerych, D.; Del Sal, G.; Paci, P.; Bossi, G.; Piaggio, G.; et al. Mutant p53 inhibits miRNA biogenesis by interfering with the microprocessor complex. Oncogene 2016, 35, 3760–3770.
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208.
- Tragante, V.; Moore, J.H.; Asselbergs, F.W. The ENCODE Project and Perspectives on Pathways. Genet. Epidemiol. 2014, 38, 275–280.
- Wang, K.C.; Chang, H.Y. Molecular Mechanisms of Long Noncoding RNAs. Mol. Cell 2011, 43, 904–914.
- Bhan, A.; Soleimani, M.; Mandal, S. Long Noncoding RNA and Cancer: A New Paradigm. Cancer Res. 2017, 77, 3965–3981.
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874.
- Huarte, M.; Guttman, M.; Feldser, D.M.; Garber, M.; Koziol, M.J.; Kenzelmann-Broz, D.; Khalil, A.M.; Zuk, O.; Amit, I.; Rabani, M.; et al. A Large Intergenic Noncoding RNA Induced by p53 Mediates Global Gene Repression in the p53 Response. Cell 2010, 142, 409–419.
- Pruszko, M.; Milano, E.; Forcato, M.; Donzelli, S.; Ganci, F.; Di Agostino, S.; De Panfilis, S.; Fazi, F.; Bates, D.O.; Bicciato, S.; et al. The mutant p53-ID4 complex controls VEGFA isoforms by recruiting lncRNA MALAT1. EMBO Rep. 2017, 18, 1331–1351.
- Wang, Q.; Jiang, H.; Ping, C.; Shen, R.; Liu, T.; Li, J.; Qian, Y.; Tang, Y.; Cheng, S.; Yao, W.; et al. Exploring the Wnt Pathway-Associated LncRNAs and Genes Involved in Pancreatic Carcinogenesis Driven by Tp53 Mutation. Pharm. Res. 2014, 32, 793–805.
- Di Agostino, S.; Valenti, F.; Sacconi, A.; Fontemaggi, G.; Pallocca, M.; Pulito, C.; Ganci, F.; Muti, P.; Strano, S.; Blandino, G. Long Non-coding MIR205HG Depletes Hsa-miR-590-3p Leading to Unrestrained Proliferation in Head and Neck Squamous Cell Carcinoma. Theranostics 2018, 8, 1850–1868.
- Lin, T.; Hou, P.-F.; Meng, S.; Chen, F.; Jiang, T.; Li, M.-L.; Shi, M.-L.; Liu, J.-J.; Zheng, J.; Bai, J. Emerging Roles of p53 Related lncRNAs in Cancer Progression: A Systematic Review. Int. J. Boil. Sci. 2019, 15, 1287–1298.
- Zhao, Y.; Li, Y.; Sheng, J.; Wu, F.; Li, K.; Huang, R.; Wang, X.; Jiao, T.; Guan, X.; Lu, Y.; et al. P53-R273H mutation enhances colorectal cancer stemness through regulating specific lncRNAs. J. Exp. Clin. Cancer Res. 2019, 38, 379.
- Ji, P.; Diederichs, S.; Wang, W.; Boing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041.
- Yamada, K.; Kano, J.; Tsunoda, H.; Yoshikawa, H.; Okubo, C.; Ishiyama, T.; Noguchi, M. Phenotypic characterization of endometrial stromal sarcoma of the uterus. Cancer Sci. 2006, 97, 106–112.
- Guffanti, A.; Iacono, M.; Pelucchi, P.; Kim, N.; Soldà, G.; Croft, L.; Taft, R.J.; Rizzi, E.; Askarian-Amiri, M.; Bonnal, R.; et al. A transcriptional sketch of a primary human breast cancer by 454 deep sequencing. BMC Genom. 2009, 10, 163.
- Gutschner, T.; Hämmerle, M.; Diederichs, S.; Haemmerle, M. MALAT1 — A paradigm for long noncoding RNA function in cancer. J. Mol. Med. 2013, 91, 791–801.
- Bernard, D.; Prasanth, K.V.; Tripathi, V.; Colasse, S.; Nakamura, T.; Xuan, Z.; Zhang, M.Q.; Sedel, F.; Jourdren, L.; Coulpier, F.; et al. A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. EMBO J. 2010, 29, 3082–3093.
- Chen, R.; Liu, Y.; Zhuang, H.; Yang, B.; Hei, K.; Xiao, M.; Hou, C.; Gao, H.; Zhang, N.; Jia, C.; et al. Quantitative proteomics reveals that long non-coding RNA MALAT1 interacts with DBC1 to regulate p53 acetylation. Nucleic Acids Res. 2017, 45, 9947–9959.
- Wolfgang, C.L.; Herman, J.M.; Laheru, D.A.; Klein, A.P.; Erdek, M.A.; Fishman, E.K.; Hruban, R.H. Recent progress in pancreatic cancer. CA A Cancer J. Clin. 2013, 63, 318–348.
- Cordani, M.; Oppici, E.; Dando, I.; Butturini, E.; Pozza, E.D.; Nadal-Serrano, M.; Oliver, J.; Roca, P.; Mariotto, S.; Cellini, B.; et al. Mutant p53 proteins counteract autophagic mechanism sensitizing cancer cells to mTOR inhibition. Mol. Oncol. 2016, 10, 1008–1029.
- Fiorini, C.; Cordani, M.; Padroni, C.; Blandino, G.; Di Agostino, S.; Donadelli, M. Mutant p53 stimulates chemoresistance of pancreatic adenocarcinoma cells to gemcitabine. Biochim. et Biophys. Acta BBA Bioenerg. 2015, 1853, 89–100.
- Morton, J.P.; Timpson, P.; Karim, S.A.; Ridgway, R.A.; Athineos, D.; Doyle, B.; Jamieson, N.B.; Oien, K.A.; Lowy, A.M.; Brunton, V.G.; et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2009, 107, 246–251.
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The Mutational Landscape of Head and Neck Squamous Cell Carcinoma. Science 2011, 333, 1157–1160.
- Network, T.C.G.A.; Network, C.G.A. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582.
- Der Plas, M.L.-V.; Brakenhoff, R.H.; Kuik, D.J.; Buijze, M.; Bloemena, E.; Snijders, P.J.; Leemans, C.R.; Braakhuis, B.J. Prognostic Significance of Truncating TP53 Mutations in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2011, 17, 3733–3741.


