 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Víctor M. Víctor | + 4552 word(s) | 4552 | 2021-05-25 05:37:41 | | | |
| 2 | Karina Chen | Meta information modification | 4552 | 2021-06-03 11:57:52 | | |
Video Upload Options
Emerging evidence reveals that oxidative stress has a critical role in the development of type 2 diabetes. Overproduction of reactive oxygen species can promote an imbalance between the production and neutralization of antioxidant defence systems, thus favoring lipid accumulation, cellular stress, and the activation of cytosolic signaling pathways, and inducing β-cell dysfunction, insulin resistance, and tissue inflammation. Over the last few years, microRNAs (miRNAs) have attracted growing attention as important mediators of diverse aspects of oxidative stress. These small endogenous non-coding RNAs of 19–24 nucleotides act as negative regulators of gene expression, including the modulation of redox signaling pathways.
1. Oxidative Stress and Diabetes
Oxidative stress, defined as elevated intracellular levels of reactive oxygen species (ROS) and decreased in antioxidant content, plays a key role in the onset of diabetes and its related disorders [1]. ROS are short-living, highly bioactive molecules resulting from the reduction of molecular oxygen. They include hydroxyl radical, hydrogen peroxide, superoxide, hypochlorous acid, peroxynitrite, and lipid radicals, among others, all of which serve as secondary messengers in intracellular signaling of different biological processes (cell-cell adhesion and signaling, cell differentiation, development, and cell death).
The main sources of ROS are mitochondria, organelles that host critical metabolic processes such as β-oxidation of fatty acids, adenosine triphosphate (ATP)-generating oxidative phosphorylation (OXPHOS), biosynthetic pathways, and mediation of cell death and calcium chelation, amongst many others [2]. Their primary function is oxidative energy production, which takes place at the electron transport chain (ETC), a sequence of four electron transfer complexes (I–IV) integrated in the mitochondrial inner membrane [3]. This process begins with the appearance of NADH and FADH2 as a result of two main routes of energy, the Krebs cycle, and β-oxidation of fatty acids, which shuttle high energy molecules to the ETC. NADH and FADH2 interact with ETC complex I and II, respectively. This electron transfer generates a proton gradient between the mitochondrial matrix and the intermembrane space; in turn, the energy accumulated is used by complex V to synthesize ATP. Even though most O2 is completely consumed during OXPHOS, a small part (1–2%) becomes superoxide anion (O2−) at complexes I and III. Under physiological conditions, ROS production and degradation are balanced through antioxidant defence mechanisms, including dismutase (SOD), glutathione peroxidase (GPx), catalase, and the (Keap1)-NRF2-ARE pathway [4]. However, during an inappropriate compensatory response, ROS generation is enhanced considerably in response to different stimuli, thus activating intracellular stress-associated pathways. Notably, accumulating evidence suggests that hyperglycemia, as well as hyperlipidemia, can contribute to low-grade inflammation and oxidative stress, both of which are potentially harmful to healthy cells, since they contribute to central processes that are interrupted during diabetes (insulin secretion, insulin action, or both) [5].
Several pathways have been proposed to explain how hyperglycemia and specifically the metabolic state of diabetes generate damage and oxidative stress in cells and tissues requiring (or not) insulin for efficient glucose uptake. These include autoxidation of glucose, protein kinase C (PKC) dependent activation of NAD(P)H oxidase, increased glycolysis, intercellular activation of the sorbitol (polyol) pathway, an augmented hexosamine pathway, increased intracellular formation of AGEs, and enhanced expression of the receptor for AGEs (RAGE) [6]. The activation of these pathways due to excessive production of ROS and prolonged hyperglycemia contributes to the inhibition of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and, in turn, to the accumulation of specific precursors of the glycolytic pathway, such as fructose-6-phosphate or glyceraldehyde-3-phosphate. In this regard, the subsequent activation of the polyol pathway causes NADPH depletion, thus contributing to oxidative stress and reducing intracellular levels of glutathione, one of the major intracellular antioxidants [7][8].
It is important to mention that, after prolonged exposure to high glucose levels, other ROS sources are activated, among them AGEs. AGEs are heterogeneous products generated by non-enzymatic glycosylation of lipids or proteins during the hyperglycemic state, and they elicit their function by binding to their receptor, RAGE [9]. This interaction activates the nuclear transcription factor kappa-B (NF-κB) pathway and NADPH oxidase, thus promoting oxidative stress and inflammation, and contributing to diabetic vascular complications [10][11]. In addition, AGEs have been shown to neutralize NO, reduce endothelial NO synthase (eNOS) activity, stimulate cell adhesion molecule expression [12], and increase the potent vasoconstrictor endothelin-1 (ET-1), thus affecting endothelial function and altering the structural integrity of the vascular wall [13]. Growing evidence also suggests that excessively high AGEs are linked to β-cell damage and peripheral IR through a variety of mechanisms, including inhibition of cytochrome-c oxidase and reduction of ATP production [14], impairment of mitochondrial function [15], generation of oxidative stress, and induction of inflammatory events [16]. Regarding the last of these mechanisms, studies performed in human umbilical vein endothelial cells (HUVECs) reveal that AGEs excreted by activated macrophages can increase the expression of proinflammatory mediators such as tumor necrosis factor-α (TNF-α), IL-1β, and IL-6 [9], and further induce mitochondrial dysfunction and cell death [17]. Specifically, AGEs induce the macrophage secretion of HMGB1 and S100, proteins that regulate endothelial cell activation, inflammation, differentiation, proliferation and cell migration, mostly via ERK1/2 and NF-κB activation [18][19]. Accumulating evidence also confirms that AGEs affect insulin secretion as well as insulin gene transcription. For example, Zhao et al. showed that AGEs inhibit ATP production and cytochrome c oxidase activity in murine pancreatic islets, thus inducing iNOS expression and impairing insulin secretion, which leads to an increase in blood glucose concentrations [14]. In this line, Puddu et al. reported that the decreased insulin content observed in the clonal β-cell line HIT-T15 after AGEs treatment is associated with decreased expression of PDX-1 (pancreatic and duodenal homeobox 1) and an increase of FoxO1 (Forkhead box protein O1), both of which play key roles in β-cell maturation, as well as in gluconeogenesis and glycogenolysis. Subsequent research has suggested that AGEs also degrade pancreatic β-cells, thus contributing to their impaired function or apoptosis. Studies by Lim et al. [20] showed that AGEs treatment induces ROS formation and RAGE expression, as well as causing β-cells apoptosis. Antioxidant treatment and RAGE inhibition prevented these changes, suggesting that AGEs stimulate cell death through RAGE-induced ROS generation. Accordingly, Lin et al. reported an apoptotic morphology in AGE-treated INS-1 cells (a well-established model for studies of pancreatic islet beta-cell function). These effects are due to AGE-induced oxidative stress generated through both stress-related signaling pathways (p38 and Jun N-terminal kinase) and the mitochondrial electron transport chain, which activate ROS production via NAPDH oxidase [21]. Although AGEs are principally produced endogenously, they can also be derived from diet (e.g., meat, cheese, coffee, milk), especially from food stored for long periods, prepared under high temperature conditions, or containing additives. Therefore, preventing dietary uptake of AGEs and endogenous AGEs formation may represent an integral part of comprehensive diabetes care.
In recent years, DAG–protein kinase (PK)C activation has received increasing attention as another critical pathway that links hyperglycemia to oxidative stress and the diabetic complications related to it. While DAG is a second-messenger signaling lipid, PKC is a serine/threonine-related protein kinase associated with vascular alterations such as angiogenesis, permeability, defects in extracellular matrix synthesis, cytokine activation and inhibition, cell growth and apoptosis, and leukocyte adhesion [22]. It is widely accepted that high glucose levels activate the formation of DAG, which, in turn, binds to PKC and causes its activation and translocation to the plasma membranes. PKC phosphorylates NADPH oxidase, thereby stimulating, either directly or indirectly, the generation of superoxide and further promoting oxidative stress [23]. Consequently, activation of the DAG/PKC pathway leads to phosphorylation and reduced activity of eNOS, along with an increment of platelet aggregation and vasoconstriction via ET-1 production. In addition, this pathway has been shown to contribute to the inflammatory process by activating adhesion molecules and cytokines [6][23][24]. Interestingly, PKC activation has also been shown to inhibit downstream metabolic enzymes and different insulin signaling cascade components [25]. Indeed, PKC seems to directly phosphorylate serine residues of the insulin receptor substrates, especially IRS-1, and promote their degradation, thus attenuating insulin signaling and inducing IR [26].
Of note, pancreatic β-cells are particularly vulnerable to oxidative stress, as they express relatively low levels of some antioxidant systems and peroxide-metabolizing enzymes (i.e., GSH peroxidase, catalase, and SOD). Under physiological conditions, β-cells release insulin in response to blood glucose levels. However, conditions like hyperglycemia, increased metabolic stress, and peripheral IR can lead to mitochondrial dysfunction, activation of the mitochondrial cytochrome c-mediated apoptotic pathway, and enhanced ROS generation. The oxidative stress generated can directly damage β-cells by oxidizing lipids, proteins, and DNA, which causes their dysfunction and/or death through several processes such as changes in dysregulated gene expression, receptor signal transduction, enzymatic activity, ion channel transport, and apoptosis [27][28]. Consequently, both insulin action and production by β-cells become deficient.
Excessive ROS levels can also indirectly damage β-cells by activating different stress-sensitive intracellular signaling pathway mediators such as p38 mitogen-activated protein kinases (p38 MAPK), NF-κB, c-Jun N-terminal kinase/stress activated protein kinases (JNK/SAPK), among others. These changes can reduce mitochondrial ATP production by down-regulating respiratory chain proteins, thus impairing insulin production [29]. Several mitochondrial pathways are reported to be implicated in ROS production in β-cells under hyperglycemia, including increased intracellular AGE production, oxidative phosphorylation, protein kinase C (PKC) activation, and polyol pathway activation, which have been mentioned previously.
Regarding pancreatic β-cell function and survival, oxidative stress interferes with three essential pathways: c-Jun N-terminal kinase (JNK) activation, AMP-activated protein kinase (AMPK) activation, and mammalian target of rapamycin (mTOR) inhibition. The AMPK pathway regulates several β-cell processes, including proliferation, insulin secretion, and survival. Under normal conditions, glucose stimulation leads to a reduction in AMPK phosphorylation and related activation; however, this reduction is markedly attenuated in pathological states [30]. In vivo studies performed by Zhang et al. demonstrated that ROS-mediated overexpression of pAMPK increases extracellular-signal-regulated kinase (pERK), which is involved in reduced β-cell mass and impaired β-cell proliferation [31]. Moreover, pAMPK up-regulation increased β-cell death and reduced insulin production in murine pancreatic cells. Interestingly, pAMPK may have an inhibitory effect on mTOR, a nutrient-responsive serine-threonine kinase that plays a crucial role in increasing and maintaining β-cell mass by regulating autophagy, cell growth, translation, cell size, apoptosis, and proliferation [32]. Indeed, it is known that, under oxidative stress, mTOR is inhibited through the activation of AMPK. Thus, mTOR inactivation leads to several detrimental effects in multiple downstream intracellular processes, among which increased expression of the thioredoxin-interacting protein (TXNIP) is the most relevant [33][34]. TXNIP is a ubiquitously expressed protein that negatively modulates the TXN antioxidant systems, influencing cellular antiapoptotic and antioxidant mechanisms [35]. In this regard, several studies have confirmed that, once inside the mitochondria, TXNIP binds to TXN2 and initiates mitochondria-mediated β-cell apoptosis through the apoptosis-signal-regulating kinase, namely ASK1 [36]. Of note, TXNIP-induced apoptosis is associated with activation of the NLR family pyrin domain containing 3 (NLRP3) inflammasome, a multimeric protein complex known to influence the innate immune system. Particularly, TXNIP induces NLRP3 inflammasome assembly, which recruits procaspase-1 to generate active caspase-1 and then converts the immature cytokines pro-IL-18 and pro-IL-1β into their mature forms, IL-18 and IL-1β. These activated cytokines contribute to the subsequent inflammatory effect, thus mediating oxidative stress-induced pancreatic islet dysfunction [37].
Another essential pathway activated in β-cells under oxidative stress conditions is the JNK pathway. JNK activation is involved in promoting impaired insulin signaling and apoptosis through serine phosphorylation and further inactivation of insulin receptor substrate 1/2 (IRS1/2) and the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) pathway [38][39]. Concurrently, inactivation of the PI3K/AKT pathway leads to down-regulation of mTOR and the subsequent loss of β-cell mass, as well as the nuclear translocation of FOXO1 and reduction of PDX-1 [39], which ultimately stunts β-cell growth and proliferation [40].
In light of the reported knowledge, hyperglycemia and oxidative stress appear to be involved in different signaling pathways that contribute to β-cell dysfunction, tissue inflammation, and insulin resistance (Figure 1). Therefore, preservation of glucose and redox homeostasis is essential to prevent diabetes. In this regard, microRNAs (miRNAs, miRs) have become the focus of increasing interest as important mediators in regulating diverse aspects of the cellular oxidative stress typical of diabetic conditions.
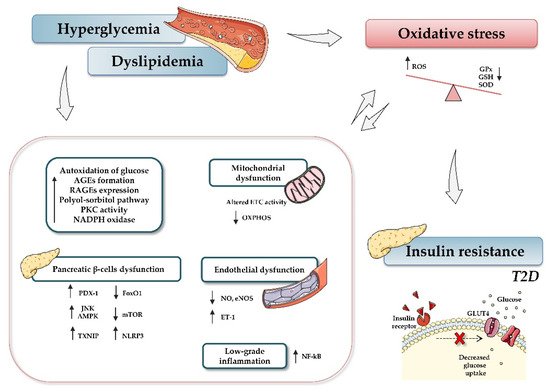
Figure 1. Relationship between type 2 diabetes and oxidative stress. Hyperglycemia, dyslipidemia, and insulin resistance play key roles in oxidative stress and diabetes development. Briefly, increased glucose levels promote different mechanisms, such as autoxidation of glucose, accumulation of advanced glycation-end products (AGE) and nitric oxide (NO), activation of diacylglycerol (DAG), activation of polyol-sorbitol pathway, and an increase in protein kinase C (PKC), which, in turn, lead to the generation of oxidative stress, impairment of mitochondrial function, and induction of inflammatory events. The figure summarizes the most relevant involved processes and signaling pathways. Up and down arrows indicate an increase and decrease, respectively. Abbreviations: AGEs, advanced glycation-end products; AMPK, AMP-activated protein kinase; ETC, electron transport chain; FoxO1, Forkhead box protein O1; GPx, glutathione peroxidase; GSH, reduced glutathione; NADPH, nicotinamide adenine dinucleotide phosphate; NF-кB, nuclear factor kappa-light-chain-enhancer of activated B cells; NO, nitric oxide; eNOS, endothelial nitric oxide synthase; mTOR, mammalian target of rapamycin; NLRP3, (nucleotide oligomerization domain (NOD), leucine-rich repeat (LRR) and pyrin domain (PYD)); OXPHOS, oxidative phosphorylation system; PDX-1, pancreatic and duodenal homeobox 1; PKC, protein kinase C; RAGEs, receptor for AGEs; ROS, reactive oxygen species; SOD, dismutase; JNK, c-Jun N-terminal kinase; T2D, type 2 diabetes; TXNIP, thioredoxin-interacting protein.
2. MiRNAs
MiRNAs are small (20–25 nt) non-coding, single stranded RNAs that inhibit gene expression at the transcriptional or post-transcriptional level. They are known to target multiple genes, and, conversely, genes are targeted by multiple miRNAs, thus allowing many levels of regulation of gene expression. MiRNAs originate from a double-stranded RNA transcript folded in a hairpin structure, known as pre-miRNA precursor molecules. The canonical biogenesis of miRNAs requires two central steps; in the nucleus, the enzyme Drosha, in association with the double-stranded RNA-binding protein DGCR8, crops pre-miRNA to a 70-nucleotide (nt) pre-miRNA, which is then transported to the cytosol. There, pre-miRNA undergoes further processing by the Dicer-TRBP (transactivation-responsive RNA-binding protein) complex into a miRNA duplex approximately 20 nt long, with a 3′- strand and a 5′- terminus strand. Although both arms can become functional, one strand is typically degraded and the other is selected as mature miRNA, depending on the cell type or the tissue. Subsequently, this single-stranded miRNA directly interacts with proteins of the Argonaute (AGO) family, components of “miRNA-induced silencing complexes” (miRISCs). These effector complexes promote mRNA deadenylation, translational repression or endonucleolytic cleavage of highly complementary targets, and exonucleolytic decay of partially complementary ones [41]. Besides this canonical miRNA biogenesis, illustrated in Figure 2, an alternative non-canonical pathway that bypasses Drosha or Dicer processing has been described [42]. miRISC, through sequence-specific binding to the UTR of target mRNAs, is able to guide the miRNA to the specific target, thus interfering with translational initiation factors and translation and protein synthesis, as well as blocking translation post-initiation [42].
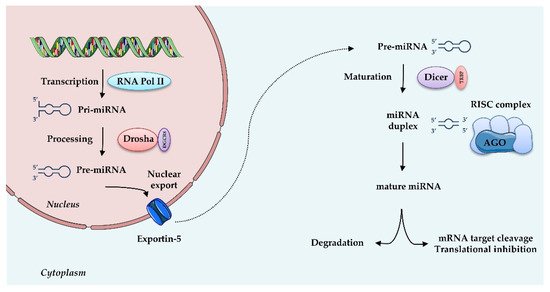
Figure 2. MicroRNAs biogenesis: microRNAs are transcribed by RNA Polymerase II (RNA Pol II) as large RNA precursors named pri-miRNA. Pri-miRNA is processed by the enzyme Drosha in association with the RNA-binding protein DGCR8 (DiGeorge syndrome critical region gene 8), which excises the stem-loop to form pre-miRNA. Pre-miRNAs are exported from the nucleus by a carrier protein known as Exportin-5. In the cytoplasm, pre-miRNA undergoes further processing by the Dicer-TRBP complex into a miRNA duplex approximately 20 nt long, known as 3′ and 5′ end strands. Typically, the first strand is degraded and the second is selected as mature miRNA. Mature miRNA is loaded into RNA induced silencing complex (RISC) via the AGO proteins and is then able to regulate gene expression by translational repression or induction of mRNA degradation. Increasing evidence suggests that, depending on the cell type or tissue, both strands can generate functional mature miRNAs.
It is thought that miRNAs modulate the expression of up to 60% of the human genome’s genes [43], and many of them have been shown to participate in the development of diabetes. Particularly, several dysregulated miRNAs from insulin-sensitive organs, including insulin-producing pancreatic β-cells, white adipose tissue, and skeletal muscle, have been related to various diabetes-associated processes, including insulin secretion, endothelial dysfunction, adipocyte differentiation, and pancreatic β-cells [44][45].
3. Oxidative Stress, miRNAs, and Diabetes
This oxidative stress has been implicated in several pathological mechanisms of diabetes: deregulation of antioxidant pathways, inflammation, altered glycemic control, changes in lipid metabolism and structure, protein oxidation, and mitochondrial and β-cell dysfunction. Therapeutic approaches aimed at reducing oxidative stress may offer protection against diabetes and its complications. However, antioxidant treatments have mostly proved unsuccessful to date [46].
Multiple miRNAs have been evaluated in cellular and animal models of diabetes, as well as in human samples, and we aim to provide an overview of the latest and most important ones.
3.1. MiRNAs, Hyperglycemia-Induced ROS and Endothelial Dysfunction
A recent report has shown that patients with T2D and impaired glucose tolerance present increased miR-21 plasma levels [47], which leads to an undermined antioxidant response and disruption of ROS homeostasis. Indeed, miRNA-21 inhibition was able to reverse the effect of its putative ROS-homeostatic target genes, such as FoxO1 and SOD2 [48], whose expression is diminished under diabetic conditions. These findings were confirmed by an in vivo experiment in mice in which miR-21 suppression stimulated the nuclear peroxisome proliferator activated receptor (PPAR) [49], known to be an important receptor in the regulation of homeostasis, and the metabolism of glucose and lipids [50]. In this context, many studies have demonstrated the crucial role of the miR-200 family in the oxidative stress and endothelial inflammation present in diabetes and its associated diseases [51]. The miR-200 family includes different evolutionarily conserved miRNAs (miR-429, miR-141, miR-200a, miR-200b, and miR-200c) implicated in the regulation of redox imbalance and positively modulated by H2O2. In particular, it has been found that overexpression of miR-200c reduces endothelial cell growth in HUVECs, thus inducing their senescence and apoptosis, while inhibition of the same miRNA leads to the restoration of endothelial function [51]. The endothelial function has been seen to take place via up-regulation of zinc finger E-box-binding homeobox (ZEB1) [51], a homeodomain transcription factor that protects against oxidative stress-associated endothelial dysfunction. Similarly, Belgardt et al. revealed that mice treated with streptozotocin (STZ), an agent that causes DNA damage, oxidative stress, and subsequently diabetes, displayed overexpression of miR-200 in pancreatic β-cells. In contrast, miR-200–silenced mice exhibited unaltered β-cell function and normal metabolic parameters, and were partially protected from STZ-induced hyperglycemia [52]. Interestingly, in vitro findings have demonstrated that miR-200 can also regulate sirtuin 1 (SIRT1) [53], a class III histone deacetylase that acts as a sensor of oxidative stress and a modulator of several cellular mechanisms, including insulin resistance and energy balance. Generally, SIRT-1 cooperates with AMPK in order to activate FOXOs, peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) and PPARα, and, in turn, antioxidant signals, to suppress stress. By targeting SIRT1, miR-200 decreases NO and increases the acetylation of SIRT1 targets, thus causing ROS generation and endothelial dysfunction. Subsequent in vivo studies have confirmed these data, highlighting that elevated ROS production occurs through the targeting and negative regulation of peroxiredoxin 2 (PRDX2) [53], a redox protein that contributes to the antioxidant defence system. Of note, SIRT1 is targeted by other miRNAs, thus increasing diabetes-associated oxidative stress. Among these miRNAs, miR-34, miR-204, and miR-106b can be counted [54]. MiR-34 downregulates SIRT1, causing hyperglycemia-induced vascular cell senescence [55]; miR-204 promotes endothelial dysfunction and vascular ER stress [56]; and miR-106b overexpression increases the oxidative stress induced by high glucose levels in NIT-1 cells, a mouse pancreatic β-cell line [57].
3.2. MiRNAs and AGE-Induced Oxidative Stress
As previously mentioned, prolonged exposure to high glucose levels can generate AGEs, which activate several signaling pathways that elicit ROS production, NO activity, and chronic and acute diabetes-related inflammatory processes. In this line, several miRNAs are reported to play an essential role in AGE modulation. Li et al. [58] observed significant up-regulation of miR-214 in human AGE-treated THP-1 monocytes, which delayed their apoptotic processes and acted as a molecular signature of chronic inflammation. A similar up-regulation of miR-214 was also detected in peripheral monocytes from patients with chronic renal failure. Interestingly, using bioinformatics analysis, the authors provided a list of predicted miR-214 target genes, among which the most relevant was PTEN, a pro-apoptotic molecule that normally acts on AKT [59]. Upon overexpression of miR-214, the amount of PTEN decreases and is unable to activate Akt, thus contributing to monocyte growth and survival and the inflammation that subsequently occurs [58]. In addition, studies performed in human renal tubular cells have indicated that AGE-induced oxidative and ER stress reduces miR-205 and induces ROS generation by reducing heme oxygenase-1 (HO-1), SOD-1 and SOD-2 [60]. These findings suggest that miR-205 improves antioxidant enzyme expression, and that its down-regulation results in the impairment of cell survival under stress. Moreover, up-regulation of miR-21 has been related to oxidative stress in that it reduces NO bioavailability and increases intracellular ROS levels via SOD2 targeting [61]. Of special relevance to the AGE-induced oxidative stress axis is miR-200b/miR-200c. Loss of miR-200b/miR-200c function in AGE-treated endothelial cells has been shown to induce ROS production and apoptosis via up-regulation of RhoA/RhoA/Rho associated kinase 2 (ROCK2) signaling [62]. ROCK is a serine/threonine kinase that interacts with activated Rho GTPases, thus controlling several critical cell processes, such as proliferation, differentiation, motility, adhesion, apoptosis, and ROS generation. Wu et al. provided evidence that the ROCK signaling pathway is an essential player in oxidative stress-induced damage in HUVECs. The increased cell permeability they observed was partially due to F-actin depolymerization in response to AGEs. Reduction of RhoA/ROCK2 signaling by miR-200c and miR-200b mimics led to F-actin remodeling and a further amelioration of AGE-induced injury. Unfortunately, the exact mechanism responsible for this is unclear, and further studies are warranted to clarify it.
3.3. MiRNAs and Pancreatic β-Cell Function
Accumulating evidence also suggests that miRNAs are key players in pancreas β-cell differentiation and function. Among them, miR-375, a pancreatic islet-specific miRNA, has been involved in the final stages of insulin secretion by regulating myotrophin expression in cultured MIN6 cells [63], a pancreatic β-cell line. Myotrophin is a cytoplasmic protein involved in insulin granule exocytosis. Consequently, miR-375 overproduction and the reduced expression of its target myotrophin attenuate insulin release [63]. Based on these data, further murine studies were performed, revealing that the specific knockdown of miR-375 led to reduced β-cell mass and proliferation, increased glucagon release from isolated islets, and subsequent hepatic gluconeogenesis and glucose production [64]. The process by which loss of miR-375 function directs to reduced cell mass is currently unknown and requires further research.
Interestingly, many researchers underline the important role of miRNAs in controlling β-cell insulin content through the downregulation of several transcriptional repressors. Melkman-Zehava et al. demonstrated that specific suppression of miR-182, miR-148, miR-26, or miR-24 in isolated primary islets or cultured MIN6 cells downregulates insulin mRNA levels and insulin promoter activity [65]. Indeed, knockdown of the above mentioned miRNAs has been shown to significantly upregulate Sox6 and Bhlhe22, two target genes known to repress insulin expression [66].
Similar to miR-24, miR-26, miR-148, and miR-182, miR-30d is a positive regulator of insulin transcription. Specifically, it downregulates mitogen-activated protein 4 kinase 4 (MAP4K4), a cytokine-inducible kinase that, when activated, inhibits insulin production and release [67]. In this regard, a reduction of miR-30d has been detected in islets isolated from diabetic mice, in which the expression of MAP4K4 was markedly augmented. The same study revealed that miR-30d induces insulin gene transcription by activating MafA (v-maf musculoaponeurotic fibrosarcoma oncogene homolog A), a transcription factor that regulates β-cell activity, affecting β-cell mass, insulin transcription, and insulin secretion. Several reports have indicated that the sustained expression of MafA results in augmented β-cell mass, higher plasma insulin levels, and significantly lower plasma glucose levels [68], thus painting a picture of miR-30d as a promising therapeutic target for diabetes management.
Of particular relevance in this sense is miR-7, a negative regulator of insulin secretion in β-cells. Latreille et al. showed that miR-7 regulates β-cell function by directly targeting genes that regulate glucose-stimulated insulin secretion [69], and its genetic deletion improves glucose tolerance in mice. Similarly, using transgenic mice, they demonstrated that overexpression of miR-7 leads to β-cell dedifferentiation, impaired insulin release and, subsequently, diabetes [69]. Lastly, the same authors analyzed miR-7 levels in islets from mildly T2D, non-diabetic obese, and control human subjects. As expected, miR-7 levels were lower in non-diabetic and obese individuals, thus confirming the results obtained in mouse models. Surprisingly, T2D patient islets also displayed lower miR-7 levels than healthy controls, thus denoting a transient phase of the disease immediately prior to a prediabetes condition [69].
In the last few years, miR-7a, the most abundant form of miR-7 in islet cells, has been shown to inhibit mTORC signaling, an intracellular pathway that coordinates insulin signaling by regulating different components, such as insulin-like growth factor 1 receptor/insulin receptor (IGF-IR/IR), insulin receptor substrate (IRS-1), growth factor receptor-bound protein 10 (Grb10), and F-box/WD repeat-containing protein 8 (Fbw8), among others [70]. MiR-7 seems to act on five of its main components: two MAPK-interacting kinases, Mknk1 and Mknk2; two main downstream effectors of eukaryotic translation initiation factor 4E (eIF4E), p70S6K and TORC1; and one of the essential TORC2 components, Mapkap1. Wang et al. also demonstrated that silencing miR-7a in mouse primary islets can activate mTOR signaling and promote β-cell replication and proliferation [71]. For this reason, miR-7 is one of the most extensively evaluated RNA regulators and potentially attractive therapeutic targets in diabetes, since it represents a “brake” for β-cell proliferation.
Like miR-7, miR-9 exerts a negative control on glucose-induced insulin release in β-cells. In this regard, Roggli et al. suggested that overexpression of miR-9 in MIN6 and dissociated islet cells is responsible for defective insulin release by directly targeting the transcription factor Onecut2. This factor is known to bind to the granuphilin promoter, a potent inhibitor of insulin exocytosis [72], and to repress its expression and transcriptional activity, thus undermining the secretion of insulin. In addition, Roggli et al. demonstrated that overexpression of Onecut2 alters insulin secretion in INS-1E cells, a widely used β-cell model, suggesting that excessive levels of Onecut2 can be deleterious for β-cell function.
3.4. MitomiR
As reported in some of our previous reviews and original studies [73][74], mitochondrial dysfunction associated with oxidation of fatty acids is closely linked to oxidative stress and the further onset of insulin resistance and T2D. Notably, some recently discovered miRNAs, known as mitochondrial-located miRNAs (MitomiR), have been shown to directly alter mitochondrial functions. In this sense, they seem to interact with mitochondrial genome-derived mRNA molecules and further regulate several proteins that are critical for these organelles’ metabolism, as well as for OXPHOS and lipid metabolism [75][76]. Studies performed in skeletal muscle from transgenic mice showed that overexpression of miR-23a leads to a reduction in PGC-1α and undermined mitochondrial function and biogenesis. Similarly, Junaith et al. reported that high fat diet (HFD)-induced mitochondrial dysfunction in skeletal muscle is associated with miR-149 down-regulation, which interferes with the SIRT-1/PGC-1α pathway [77]. The authors demonstrated that miR-149 inhibits poly(ADP-ribose) polymerase-2 (PARP-2) and increases cellular NAD+ levels and SIRT-1 activity, which further enhances mitochondrial biogenesis and function through PGC-1α activation. Indeed, it is known that some miRNAs modulate mitochondrial energy metabolism and OXPHOS by targeting various of the main mitochondrial components implicated in ETC, ATP production, and electron transfer; for example, miR-141 downregulates complex V in the mitochondrial ETC; miR-338, miR-210, and miR-181c, downregulate complex IV; and miR-210 downregulates complex III [78][79].
In light of the above-mentioned information, oxidative stress seems to be crucial for the regulation and deregulation of miRNA networks, and vice versa, as miRNA acts as a main player in different physiological and pathological events, making both of them attractive therapeutic targets.
References
- Ighodaro, O.M. Molecular pathways associated with oxidative stress in diabetes mellitus. Biomed. Pharmacother. 2018, 108, 656–662.
- Siasos, G.; Paschou, S.A.; Tousoulis, D. Mitochondria and diabetes. Ann. Transl. Med. 2020, 8, 262.
- Mailloux, R.J. An update on mitochondrial reactive oxygen species production. Antioxidants 2020, 9, 472.
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-mediated cellular signaling. Oxid. Med. Cell Longev. 2016, 2016, 4350965.
- Rains, J.L.; Jain, S.K. Oxidative stress, insulin signaling, and diabetes. Free Radic. Biol. Med. 2011, 50, 567–575.
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070.
- Pitocco, D.; Tesauro, M.; Alessandro, R.; Ghirlanda, G.; Cardillo, C. Oxidative stress in diabetes: Implications for vascular and other complications. Int. J. Mol. Sci. 2013, 14, 21525–21550.
- Brubaker, P.L.; Drucker, D.J. Minireview: Glucagon-like peptides regulate cell proliferation and apoptosis in the pancreas, gut, and central nervous system. Endocrinology 2004, 145, 2653–2659.
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced glycation end products: Sparking the development of diabetic vascular injury. Circulation 2006, 114, 597–605.
- Bierhaus, A.; Hofmann, M.A.; Ziegler, R.; Nawroth, P.P. AGEs and their interaction with AGE-receptors in vascular disease and diabetes mellitus. I. The AGE concept. Cardiovas. Res. 1998, 37, 586–600.
- Yamagishi, S.; Nakamura, N.; Suematsu, M.; Kaseda, K.; Matsui, T. Advanced glycation end products: A molecular target for vascular complications in diabetes. Mol. Med. 2015, 21, S32–S40.
- Wautier, J.L.; Zoukourian, C.; Chappey, O.; Wautier, M.P.; Guillausseau, P.J.; Cao, R.; Hori, O.; Stern, D.; Schmidt, A.M. Receptor-mediated endothelial cell dysfunction in diabetic vasculopathy. Soluble receptor for advanced glycation end products blocks hyperpermeability in diabetic rats. J. Clin. Investig. 1996, 97, 238–243.
- Santilli, F.; D’Ardes, D.; Davi, G. Oxidative stress in chronic vascular disease: From prediction to prevention. Vasc. Pharmacol. 2015, 74, 23–37.
- Zhao, Z.; Zhao, C.; Zhang, X.H.; Zheng, F.; Cai, W.; Vlassara, H.; Ma, Z.A. Advanced glycation end products inhibit glucose-stimulated insulin secretion through nitric oxide-dependent inhibition of cytochrome c oxidase and adenosine triphosphate synthesis. Endocrinology 2009, 150, 2569–2576.
- Wang, X.; Yu, S.; Wang, C.Y.; Wang, Y.; Liu, H.X.; Cui, Y.; Zhang, L.D. Advanced glycation end products induce oxidative stress and mitochondrial dysfunction in SH-SY5Y cells. In Vitro Cell Dev. Biol. Anim. 2015, 51, 204–209.
- Davis, K.E.; Prasad, C.; Vijayagopal, P.; Juma, S.; Imrhan, V. Advanced glycation end products, inflammation, and chronic metabolic diseases: Links in a chain? Crit. Rev. Food Sci. Nutr. 2016, 56, 989–998.
- Li, Y.; Chang, Y.; Ye, N.; Chen, Y.; Zhang, N.; Sun, Y. Advanced glycation end products-induced mitochondrial energy metabolism dysfunction alters proliferation of human umbilical vein endothelial cells. Mol. Med. Rep. 2017, 15, 2673–2680.
- Kokkola, R.; Andersson, A.; Mullins, G.; Ostberg, T.; Treutiger, C.J.; Arnold, B.; Nawroth, P.; Andersson, U.; Harris, R.A.; Harris, H.E. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand. J. Immunol. 2005, 61, 1–9.
- Leclerc, E.; Fritz, G.; Vetter, S.W.; Heizmann, C.W. Binding of S100 proteins to RAGE: An update. Biochim. Biophys. Acta 2009, 1793, 993–1007.
- de Paula, M.L.A.; Villela, A.M.R.; Negri, M.M.; Kanaan, S.; Weide, L.d.C.C. Role of advanced glycation end products related to the onset of diabetic kidney disease complications. Clin. Biomed. Res. 2017, 37.
- Lin, N.; Zhang, H.; Su, Q. Advanced glycation end-products induce injury to pancreatic beta cells through oxidative stress. Diabetes Metab. 2012, 38, 250–257.
- Schmitz-Peiffer, C.; Biden, T.J. Protein kinase C function in muscle, liver, and beta-cells and its therapeutic implications for type 2 diabetes. Diabetes 2008, 57, 1774–1783.
- Paneni, F.; Beckman, J.A.; Creager, M.A.; Cosentino, F. Diabetes and vascular disease: Pathophysiology, clinical consequences, and medical therapy: Part I. Eur. Heart J. 2013, 34, 2436–2443.
- Domingueti, C.P.; Dusse, L.M.; Carvalho, M.; de Sousa, L.P.; Gomes, K.B.; Fernandes, A.P. Diabetes mellitus: The linkage between oxidative stress, inflammation, hypercoagulability and vascular complications. J. Diabetes Complic. 2016, 30, 738–745.
- Schmitz-Peiffer, C. Protein kinase C and lipid-induced insulin resistance in skeletal muscle. Ann. N. Y. Acad. Sci. 2002, 967, 146–157.
- Cazzolli, R.; Mitchell, T.W.; Burchfield, J.G.; Pedersen, D.J.; Turner, N.; Biden, T.J.; Schmitz-Peiffer, C. Dilinoleoyl-phosphatidic acid mediates reduced IRS-1 tyrosine phosphorylation in rat skeletal muscle cells and mouse muscle. Diabetologia 2007, 50, 1732–1742.
- Keane, K.N.; Cruzat, V.F.; Carlessi, R.; de Bittencourt, P.I., Jr.; Newsholme, P. Molecular events linking oxidative stress and inflammation to insulin resistance and beta-cell dysfunction. Oxid. Med. Cell Longev. 2015, 2015, 181643.
- Newsholme, P.; Rebelato, E.; Abdulkader, F.; Krause, M.; Carpinelli, A.; Curi, R. Reactive oxygen and nitrogen species generation, antioxidant defenses, and beta-cell function: A critical role for amino acids. J. Endocrinol. 2012, 214, 11–20.
- Kim-Muller, J.Y.; Fan, J.; Kim, Y.J.; Lee, S.A.; Ishida, E.; Blaner, W.S.; Accili, D. Aldehyde dehydrogenase 1a3 defines a subset of failing pancreatic beta cells in diabetic mice. Nat. Commun. 2016, 7, 12631.
- Szkudelski, T.; Szkudelska, K. The relevance of AMP-activated protein kinase in insulin-secreting beta cells: A potential target for improving beta cell function? J. Physiol. Biochem. 2019, 75, 423–432.
- Zhang, Y.; Yamamoto, T.; Hisatome, I.; Li, Y.; Cheng, W.; Sun, N.; Cai, B.; Huang, T.; Zhu, Y.; Li, Z.; et al. Uric acid induces oxidative stress and growth inhibition by activating adenosine monophosphate-activated protein kinase and extracellular signal-regulated kinase signal pathways in pancreatic beta cells. Mol. Cell Endocrinol. 2013, 375, 89–96.
- Blandino-Rosano, M.; Chen, A.Y.; Scheys, J.O.; Alejandro, E.U.; Gould, A.P.; Taranukha, T.; Elghazi, L.; Cras-Meneur, C.; Bernal-Mizrachi, E. mTORC1 signaling and regulation of pancreatic beta-cell mass. Cell Cycle 2012, 11, 1892–1902.
- Chau, G.C.; Im, D.U.; Kang, T.M.; Bae, J.M.; Kim, W.; Pyo, S.; Moon, E.Y.; Um, S.H. mTOR controls ChREBP transcriptional activity and pancreatic beta cell survival under diabetic stress. J. Cell Biol. 2017, 216, 2091–2105.
- Maedler, K.; Ardestani, A. mTORC in beta cells: More than only recognizing comestibles. J. Cell Biol. 2017, 216, 1883–1885.
- Alhawiti, N.M.; Al Mahri, S.; Aziz, M.A.; Malik, S.S.; Mohammad, S. TXNIP in metabolic regulation: Physiological role and therapeutic outlook. Curr. Drug Targets 2017, 18, 1095–1103.
- Saxena, G.; Chen, J.; Shalev, A. Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. J. Biol. Chem. 2010, 285, 3997–4005.
- Sokolova, M.; Sahraoui, A.; Hoyem, M.; Ogaard, J.; Lien, E.; Aukrust, P.; Yndestad, A.; Ranheim, T.; Scholz, H. NLRP3 inflammasome mediates oxidative stress-induced pancreatic islet dysfunction. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E912–E923.
- Zhao, Y.; Sun, H.; Li, X.; Zha, Y.; Hou, W. Hydroxysafflor yellow A attenuates high glucose-induced pancreatic beta-cells oxidative damage via inhibiting JNK/c-jun signaling pathway. Biochem. Biophys. Res. Commun. 2018, 505, 353–359.
- Yung, J.H.M.; Giacca, A. Role of c-Jun N-terminal kinase (JNK) in obesity and type 2 diabetes. Cells 2020, 9, 706.
- Hayes, H.L.; Moss, L.G.; Schisler, J.C.; Haldeman, J.M.; Zhang, Z.; Rosenberg, P.B.; Newgard, C.B.; Hohmeier, H.E. Pdx-1 activates islet alpha- and beta-cell proliferation via a mechanism regulated by transient receptor potential cation channels 3 and 6 and extracellular signal-regulated kinases 1 and 2. Mol. Cell Biol. 2013, 33, 4017–4029.
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233.
- Miyoshi, K.; Miyoshi, T.; Siomi, H. Many ways to generate microRNA-like small RNAs: Non-canonical pathways for microRNA production. Mol. Genet. Genom. 2010, 284, 95–103.
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in control of gene expression: An overview of nuclear functions. Int. J. Mol. Sci. 2016, 17, 1712.
- Eliasson, L.; Esguerra, J.L. Role of non-coding RNAs in pancreatic beta-cell development and physiology. Acta Physiol. 2014, 211, 273–284.
- Chen, H.; Lan, H.Y.; Roukos, D.H.; Cho, W.C. Application of microRNAs in diabetes mellitus. J. Endocrinol. 2014, 222, R1–R10.
- Golbidi, S.; Ebadi, S.A.; Laher, I. Antioxidants in the treatment of diabetes. Curr. Diabetes Rev. 2011, 7, 106–125.
- La Sala, L.; Mrakic-Sposta, S.; Tagliabue, E.; Prattichizzo, F.; Micheloni, S.; Sangalli, E.; Specchia, C.; Uccellatore, A.C.; Lupini, S.; Spinetti, G.; et al. Circulating microRNA-21 is an early predictor of ROS-mediated damage in subjects with high risk of developing diabetes and in drug-naive T2D. Cardiovasc. Diabetol. 2019, 18, 18.
- La Sala, L.; Mrakic-Sposta, S.; Micheloni, S.; Prattichizzo, F.; Ceriello, A. Glucose-sensing microRNA-21 disrupts ROS homeostasis and impairs antioxidant responses in cellular glucose variability. Cardiovasc. Diabetol. 2018, 17, 105.
- Gao, L.; Liu, Y.; Guo, S.; Xiao, L.; Wu, L.; Wang, Z.; Liang, C.; Yao, R.; Zhang, Y. LAZ3 protects cardiac remodeling in diabetic cardiomyopathy via regulating miR-21/PPARa signaling. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3322–3338.
- Dubois, V.; Eeckhoute, J.; Lefebvre, P.; Staels, B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J. Clin. Investig. 2017, 127, 1202–1214.
- Magenta, A.; Cencioni, C.; Fasanaro, P.; Zaccagnini, G.; Greco, S.; Sarra-Ferraris, G.; Antonini, A.; Martelli, F.; Capogrossi, M.C. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. 2011, 18, 1628–1639.
- Belgardt, B.F.; Ahmed, K.; Spranger, M.; Latreille, M.; Denzler, R.; Kondratiuk, N.; von Meyenn, F.; Villena, F.N.; Herrmanns, K.; Bosco, D.; et al. The microRNA-200 family regulates pancreatic beta cell survival in type 2 diabetes. Nat. Med. 2015, 21, 619–627.
- Carlomosti, F.; D’Agostino, M.; Beji, S.; Torcinaro, A.; Rizzi, R.; Zaccagnini, G.; Maimone, B.; Di Stefano, V.; De Santa, F.; Cordisco, S.; et al. Oxidative stress-induced miR-200c disrupts the regulatory loop among SIRT1, FOXO1, and eNOS. Antioxid. Redox Signal. 2017, 27, 328–344.
- Yamakuchi, M. MicroRNA Regulation of SIRT1. Front. Physiol. 2012, 3, 68.
- Li, Q.; Kim, Y.R.; Vikram, A.; Kumar, S.; Kassan, M.; Gabani, M.; Lee, S.K.; Jacobs, J.S.; Irani, K. P66Shc-induced microRNA-34a causes diabetic endothelial dysfunction by downregulating Sirtuin1. Arter. Thromb. Vasc. Biol. 2016, 36, 2394–2403.
- Kassan, M.; Vikram, A.; Li, Q.; Kim, Y.R.; Kumar, S.; Gabani, M.; Liu, J.; Jacobs, J.S.; Irani, K. MicroRNA-204 promotes vascular endoplasmic reticulum stress and endothelial dysfunction by targeting Sirtuin1. Sci. Rep. 2017, 7, 9308.
- Chen, D.L.; Yang, K.Y. Berberine Alleviates oxidative stress in islets of diabetic mice by inhibiting miR-106b expression and up-regulating SIRT1. J. Cell Biochem. 2017, 118, 4349–4357.
- Li, L.M.; Hou, D.X.; Guo, Y.L.; Yang, J.W.; Liu, Y.; Zhang, C.Y.; Zen, K. Role of microRNA-214-targeting phosphatase and tensin homolog in advanced glycation end product-induced apoptosis delay in monocytes. J. Immunol. 2011, 186, 2552–2560.
- Comer, F.I.; Parent, C.A. PI 3-kinases and PTEN: How opposites chemoattract. Cell 2002, 109, 541–544.
- Muratsu-Ikeda, S.; Nangaku, M.; Ikeda, Y.; Tanaka, T.; Wada, T.; Inagi, R. Downregulation of miR-205 modulates cell susceptibility to oxidative and endoplasmic reticulum stresses in renal tubular cells. PLoS ONE 2012, 7, e41462.
- Fleissner, F.; Jazbutyte, V.; Fiedler, J.; Gupta, S.K.; Yin, X.; Xu, Q.; Galuppo, P.; Kneitz, S.; Mayr, M.; Ertl, G.; et al. Short communication: Asymmetric dimethylarginine impairs angiogenic progenitor cell function in patients with coronary artery disease through a microRNA-21-dependent mechanism. Circ. Res. 2010, 107, 138–143.
- Wu, X.D.; Liu, W.L.; Zeng, K.; Lei, H.Y.; Zhang, Q.G.; Zhou, S.Q.; Xu, S.Y. Advanced glycation end products activate the miRNA/RhoA/ROCK2 pathway in endothelial cells. Microcirculation 2014, 21, 178–186.
- Poy, M.N.; Eliasson, L.; Krutzfeldt, J.; Kuwajima, S.; Ma, X.; Macdonald, P.E.; Pfeffer, S.; Tuschl, T.; Rajewsky, N.; Rorsman, P.; et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nature 2004, 432, 226–230.
- Poy, M.N.; Hausser, J.; Trajkovski, M.; Braun, M.; Collins, S.; Rorsman, P.; Zavolan, M.; Stoffel, M. miR-375 maintains normal pancreatic alpha- and beta-cell mass. Proc. Natl. Acad. Sci. USA 2009, 106, 5813–5818.
- Melkman-Zehavi, T.; Oren, R.; Kredo-Russo, S.; Shapira, T.; Mandelbaum, A.D.; Rivkin, N.; Nir, T.; Lennox, K.A.; Behlke, M.A.; Dor, Y.; et al. miRNAs control insulin content in pancreatic beta-cells via downregulation of transcriptional repressors. EMBO J. 2011, 30, 835–845.
- Iguchi, H.; Urashima, Y.; Inagaki, Y.; Ikeda, Y.; Okamura, M.; Tanaka, T.; Uchida, A.; Yamamoto, T.T.; Kodama, T.; Sakai, J. SOX6 suppresses cyclin D1 promoter activity by interacting with beta-catenin and histone deacetylase 1, and its down-regulation induces pancreatic beta-cell proliferation. J. Biol. Chem. 2007, 282, 19052–19061.
- Zhao, X.; Mohan, R.; Ozcan, S.; Tang, X. MicroRNA-30d induces insulin transcription factor MafA and insulin production by targeting mitogen-activated protein 4 kinase 4 (MAP4K4) in pancreatic beta-cells. J. Biol. Chem. 2012, 287, 31155–31164.
- Matsuoka, T.A.; Kaneto, H.; Kawashima, S.; Miyatsuka, T.; Tochino, Y.; Yoshikawa, A.; Imagawa, A.; Miyazaki, J.; Gannon, M.; Stein, R.; et al. Preserving Mafa expression in diabetic islet beta-cells improves glycemic control in vivo. J. Biol. Chem. 2015, 290, 7647–7657.
- Latreille, M.; Hausser, J.; Stutzer, I.; Zhang, Q.; Hastoy, B.; Gargani, S.; Kerr-Conte, J.; Pattou, F.; Zavolan, M.; Esguerra, J.L.; et al. MicroRNA-7a regulates pancreatic beta cell function. J. Clin. Investig. 2014, 124, 2722–2735.
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976.
- Wang, Y.; Liu, J.; Liu, C.; Naji, A.; Stoffers, D.A. MicroRNA-7 regulates the mTOR pathway and proliferation in adult pancreatic beta-cells. Diabetes 2013, 62, 887–895.
- Plaisance, V.; Abderrahmani, A.; Perret-Menoud, V.; Jacquemin, P.; Lemaigre, F.; Regazzi, R. MicroRNA-9 controls the expression of Granuphilin/Slp4 and the secretory response of insulin-producing cells. J. Biol. Chem. 2006, 281, 26932–26942.
- Burgos-Moron, E.; Abad-Jimenez, Z.; Maranon, A.M.; Iannantuoni, F.; Escribano-Lopez, I.; Lopez-Domenech, S.; Salom, C.; Jover, A.; Mora, V.; Roldan, I.; et al. Relationship between oxidative stress, ER stress, and inflammation in type 2 diabetes: The battle continues. J. Clin. Med. 2019, 8, 1385.
- Victor, V.M.; Rocha, M.; Herance, R.; Hernandez-Mijares, A. Oxidative stress and mitochondrial dysfunction in type 2 diabetes. Curr. Pharm. Des. 2011, 17, 3947–3958.
- Duarte, F.V.; Palmeira, C.M.; Rolo, A.P. The emerging role of MitomiRs in the pathophysiology of human disease. Adv. Exp. Med. Biol. 2015, 888, 123–154.
- Bandiera, S.; Mategot, R.; Girard, M.; Demongeot, J.; Henrion-Caude, A. MitomiRs delineating the intracellular localization of microRNAs at mitochondria. Free Radic. Biol. Med. 2013, 64, 12–19.
- Mohamed, J.S.; Hajira, A.; Pardo, P.S.; Boriek, A.M. MicroRNA-149 inhibits PARP-2 and promotes mitochondrial biogenesis via SIRT-1/PGC-1alpha network in skeletal muscle. Diabetes 2014, 63, 1546–1559.
- Bienertova-Vasku, J.; Sana, J.; Slaby, O. The role of microRNAs in mitochondria in cancer. Cancer Lett. 2013, 336, 1–7.
- Sun, W.; Zhao, L.; Song, X.; Zhang, J.; Xing, Y.; Liu, N.; Yan, Y.; Li, Z.; Lu, Y.; Wu, J.; et al. MicroRNA-210 modulates the cellular energy metabolism shift during H2O2-induced oxidative stress by repressing ISCU in H9c2 cardiomyocytes. Cell Physiol. Biochem. 2017, 43, 383–394.

