1. Oxidative Stress and Diabetes
Oxidative stress, defined as elevated intracellular levels of reactive oxygen species (ROS) and decreased in antioxidant content, plays a key role in the onset of diabetes and its related disorders [
16]. ROS are short-living, highly bioactive molecules resulting from the reduction of molecular oxygen. They include hydroxyl radical, hydrogen peroxide, superoxide, hypochlorous acid, peroxynitrite, and lipid radicals, among others, all of which serve as secondary messengers in intracellular signaling of different biological processes (cell-cell adhesion and signaling, cell differentiation, development, and cell death).
The main sources of ROS are mitochondria, organelles that host critical metabolic processes such as β-oxidation of fatty acids, adenosine triphosphate (ATP)-generating oxidative phosphorylation (OXPHOS), biosynthetic pathways, and mediation of cell death and calcium chelation, amongst many others [
17]. Their primary function is oxidative energy production, which takes place at the electron transport chain (ETC), a sequence of four electron transfer complexes (I–IV) integrated in the mitochondrial inner membrane [
18]. This process begins with the appearance of NADH and FADH
2 as a result of two main routes of energy, the Krebs cycle, and β-oxidation of fatty acids, which shuttle high energy molecules to the ETC. NADH and FADH
2 interact with ETC complex I and II, respectively. This electron transfer generates a proton gradient between the mitochondrial matrix and the intermembrane space; in turn, the energy accumulated is used by complex V to synthesize ATP. Even though most O
2 is completely consumed during OXPHOS, a small part (1–2%) becomes superoxide anion (O
2−) at complexes I and III. Under physiological conditions, ROS production and degradation are balanced through antioxidant defence mechanisms, including dismutase (SOD), glutathione peroxidase (GPx), catalase, and the (Keap1)-NRF2-ARE pathway [
19]. However, during an inappropriate compensatory response, ROS generation is enhanced considerably in response to different stimuli, thus activating intracellular stress-associated pathways. Notably, accumulating evidence suggests that hyperglycemia, as well as hyperlipidemia, can contribute to low-grade inflammation and oxidative stress, both of which are potentially harmful to healthy cells, since they contribute to central processes that are interrupted during diabetes (insulin secretion, insulin action, or both) [
20].
Several pathways have been proposed to explain how hyperglycemia and specifically the metabolic state of diabetes generate damage and oxidative stress in cells and tissues requiring (or not) insulin for efficient glucose uptake. These include autoxidation of glucose, protein kinase C (PKC) dependent activation of NAD(P)H oxidase, increased glycolysis, intercellular activation of the sorbitol (polyol) pathway, an augmented hexosamine pathway, increased intracellular formation of AGEs, and enhanced expression of the receptor for AGEs (RAGE) [
21]. The activation of these pathways due to excessive production of ROS and prolonged hyperglycemia contributes to the inhibition of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and, in turn, to the accumulation of specific precursors of the glycolytic pathway, such as fructose-6-phosphate or glyceraldehyde-3-phosphate. In this regard, the subsequent activation of the polyol pathway causes NADPH depletion, thus contributing to oxidative stress and reducing intracellular levels of glutathione, one of the major intracellular antioxidants [
22,
23].
It is important to mention that, after prolonged exposure to high glucose levels, other ROS sources are activated, among them AGEs. AGEs are heterogeneous products generated by non-enzymatic glycosylation of lipids or proteins during the hyperglycemic state, and they elicit their function by binding to their receptor, RAGE [
24]. This interaction activates the nuclear transcription factor kappa-B (NF-κB) pathway and NADPH oxidase, thus promoting oxidative stress and inflammation, and contributing to diabetic vascular complications [
25,
26]. In addition, AGEs have been shown to neutralize NO, reduce endothelial NO synthase (eNOS) activity, stimulate cell adhesion molecule expression [
27], and increase the potent vasoconstrictor endothelin-1 (ET-1), thus affecting endothelial function and altering the structural integrity of the vascular wall [
28]. Growing evidence also suggests that excessively high AGEs are linked to β-cell damage and peripheral IR through a variety of mechanisms, including inhibition of cytochrome-c oxidase and reduction of ATP production [
29], impairment of mitochondrial function [
30], generation of oxidative stress, and induction of inflammatory events [
31]. Regarding the last of these mechanisms, studies performed in human umbilical vein endothelial cells (HUVECs) reveal that AGEs excreted by activated macrophages can increase the expression of proinflammatory mediators such as tumor necrosis factor-α (TNF-α), IL-1β, and IL-6 [
24], and further induce mitochondrial dysfunction and cell death [
32]. Specifically, AGEs induce the macrophage secretion of HMGB1 and S100, proteins that regulate endothelial cell activation, inflammation, differentiation, proliferation and cell migration, mostly via ERK1/2 and NF-κB activation [
33,
34]. Accumulating evidence also confirms that AGEs affect insulin secretion as well as insulin gene transcription. For example, Zhao et al. showed that AGEs inhibit ATP production and cytochrome c oxidase activity in murine pancreatic islets, thus inducing iNOS expression and impairing insulin secretion, which leads to an increase in blood glucose concentrations [
29]. In this line, Puddu et al. reported that the decreased insulin content observed in the clonal β-cell line HIT-T15 after AGEs treatment is associated with decreased expression of PDX-1 (pancreatic and duodenal homeobox 1) and an increase of FoxO1 (Forkhead box protein O1), both of which play key roles in β-cell maturation, as well as in gluconeogenesis and glycogenolysis. Subsequent research has suggested that AGEs also degrade pancreatic β-cells, thus contributing to their impaired function or apoptosis. Studies by Lim et al. [
35] showed that AGEs treatment induces ROS formation and RAGE expression, as well as causing β-cells apoptosis. Antioxidant treatment and RAGE inhibition prevented these changes, suggesting that AGEs stimulate cell death through RAGE-induced ROS generation. Accordingly, Lin et al. reported an apoptotic morphology in AGE-treated INS-1 cells (a well-established model for studies of pancreatic islet beta-cell function). These effects are due to AGE-induced oxidative stress generated through both stress-related signaling pathways (p38 and Jun N-terminal kinase) and the mitochondrial electron transport chain, which activate ROS production via NAPDH oxidase [
36]. Although AGEs are principally produced endogenously, they can also be derived from diet (e.g., meat, cheese, coffee, milk), especially from food stored for long periods, prepared under high temperature conditions, or containing additives. Therefore, preventing dietary uptake of AGEs and endogenous AGEs formation may represent an integral part of comprehensive diabetes care.
In recent years, DAG–protein kinase (PK)C activation has received increasing attention as another critical pathway that links hyperglycemia to oxidative stress and the diabetic complications related to it. While DAG is a second-messenger signaling lipid, PKC is a serine/threonine-related protein kinase associated with vascular alterations such as angiogenesis, permeability, defects in extracellular matrix synthesis, cytokine activation and inhibition, cell growth and apoptosis, and leukocyte adhesion [
37]. It is widely accepted that high glucose levels activate the formation of DAG, which, in turn, binds to PKC and causes its activation and translocation to the plasma membranes. PKC phosphorylates NADPH oxidase, thereby stimulating, either directly or indirectly, the generation of superoxide and further promoting oxidative stress [
38]. Consequently, activation of the DAG/PKC pathway leads to phosphorylation and reduced activity of eNOS, along with an increment of platelet aggregation and vasoconstriction via ET-1 production. In addition, this pathway has been shown to contribute to the inflammatory process by activating adhesion molecules and cytokines [
21,
38,
39]. Interestingly, PKC activation has also been shown to inhibit downstream metabolic enzymes and different insulin signaling cascade components [
40]. Indeed, PKC seems to directly phosphorylate serine residues of the insulin receptor substrates, especially IRS-1, and promote their degradation, thus attenuating insulin signaling and inducing IR [
41].
Of note, pancreatic β-cells are particularly vulnerable to oxidative stress, as they express relatively low levels of some antioxidant systems and peroxide-metabolizing enzymes (i.e., GSH peroxidase, catalase, and SOD). Under physiological conditions, β-cells release insulin in response to blood glucose levels. However, conditions like hyperglycemia, increased metabolic stress, and peripheral IR can lead to mitochondrial dysfunction, activation of the mitochondrial cytochrome c-mediated apoptotic pathway, and enhanced ROS generation. The oxidative stress generated can directly damage β-cells by oxidizing lipids, proteins, and DNA, which causes their dysfunction and/or death through several processes such as changes in dysregulated gene expression, receptor signal transduction, enzymatic activity, ion channel transport, and apoptosis [
42,
43]. Consequently, both insulin action and production by β-cells become deficient.
Excessive ROS levels can also indirectly damage β-cells by activating different stress-sensitive intracellular signaling pathway mediators such as p38 mitogen-activated protein kinases (p38 MAPK), NF-κB, c-Jun N-terminal kinase/stress activated protein kinases (JNK/SAPK), among others. These changes can reduce mitochondrial ATP production by down-regulating respiratory chain proteins, thus impairing insulin production [
44]. Several mitochondrial pathways are reported to be implicated in ROS production in β-cells under hyperglycemia, including increased intracellular AGE production, oxidative phosphorylation, protein kinase C (PKC) activation, and polyol pathway activation, which have been mentioned previously.
Regarding pancreatic β-cell function and survival, oxidative stress interferes with three essential pathways: c-Jun N-terminal kinase (JNK) activation, AMP-activated protein kinase (AMPK) activation, and mammalian target of rapamycin (mTOR) inhibition. The AMPK pathway regulates several β-cell processes, including proliferation, insulin secretion, and survival. Under normal conditions, glucose stimulation leads to a reduction in AMPK phosphorylation and related activation; however, this reduction is markedly attenuated in pathological states [
45]. In vivo studies performed by Zhang et al. demonstrated that ROS-mediated overexpression of pAMPK increases extracellular-signal-regulated kinase (pERK), which is involved in reduced β-cell mass and impaired β-cell proliferation [
46]. Moreover, pAMPK up-regulation increased β-cell death and reduced insulin production in murine pancreatic cells. Interestingly, pAMPK may have an inhibitory effect on mTOR, a nutrient-responsive serine-threonine kinase that plays a crucial role in increasing and maintaining β-cell mass by regulating autophagy, cell growth, translation, cell size, apoptosis, and proliferation [
47]. Indeed, it is known that, under oxidative stress, mTOR is inhibited through the activation of AMPK. Thus, mTOR inactivation leads to several detrimental effects in multiple downstream intracellular processes, among which increased expression of the thioredoxin-interacting protein (TXNIP) is the most relevant [
48,
49]. TXNIP is a ubiquitously expressed protein that negatively modulates the TXN antioxidant systems, influencing cellular antiapoptotic and antioxidant mechanisms [
50]. In this regard, several studies have confirmed that, once inside the mitochondria, TXNIP binds to TXN2 and initiates mitochondria-mediated β-cell apoptosis through the apoptosis-signal-regulating kinase, namely ASK1 [
51]. Of note, TXNIP-induced apoptosis is associated with activation of the NLR family pyrin domain containing 3 (NLRP3) inflammasome, a multimeric protein complex known to influence the innate immune system. Particularly, TXNIP induces NLRP3 inflammasome assembly, which recruits procaspase-1 to generate active caspase-1 and then converts the immature cytokines pro-IL-18 and pro-IL-1β into their mature forms, IL-18 and IL-1β. These activated cytokines contribute to the subsequent inflammatory effect, thus mediating oxidative stress-induced pancreatic islet dysfunction [
52].
Another essential pathway activated in β-cells under oxidative stress conditions is the JNK pathway. JNK activation is involved in promoting impaired insulin signaling and apoptosis through serine phosphorylation and further inactivation of insulin receptor substrate 1/2 (IRS1/2) and the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) pathway [
53,
54]. Concurrently, inactivation of the PI3K/AKT pathway leads to down-regulation of mTOR and the subsequent loss of β-cell mass, as well as the nuclear translocation of FOXO1 and reduction of PDX-1 [
54], which ultimately stunts β-cell growth and proliferation [
55].
In light of the reported knowledge, hyperglycemia and oxidative stress appear to be involved in different signaling pathways that contribute to β-cell dysfunction, tissue inflammation, and insulin resistance (). Therefore, preservation of glucose and redox homeostasis is essential to prevent diabetes. In this regard, microRNAs (miRNAs, miRs) have become the focus of increasing interest as important mediators in regulating diverse aspects of the cellular oxidative stress typical of diabetic conditions.
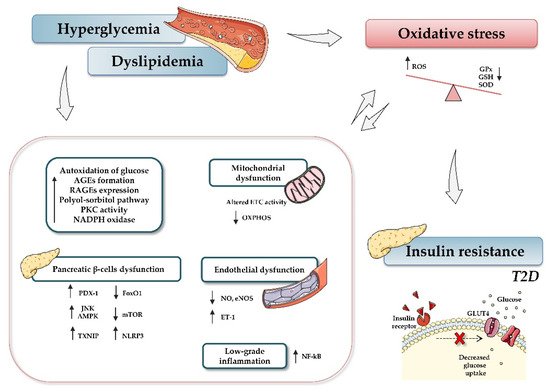
Figure 1. Relationship between type 2 diabetes and oxidative stress. Hyperglycemia, dyslipidemia, and insulin resistance play key roles in oxidative stress and diabetes development. Briefly, increased glucose levels promote different mechanisms, such as autoxidation of glucose, accumulation of advanced glycation-end products (AGE) and nitric oxide (NO), activation of diacylglycerol (DAG), activation of polyol-sorbitol pathway, and an increase in protein kinase C (PKC), which, in turn, lead to the generation of oxidative stress, impairment of mitochondrial function, and induction of inflammatory events. The figure summarizes the most relevant involved processes and signaling pathways. Up and down arrows indicate an increase and decrease, respectively. Abbreviations: AGEs, advanced glycation-end products; AMPK, AMP-activated protein kinase; ETC, electron transport chain; FoxO1, Forkhead box protein O1; GPx, glutathione peroxidase; GSH, reduced glutathione; NADPH, nicotinamide adenine dinucleotide phosphate; NF-кB, nuclear factor kappa-light-chain-enhancer of activated B cells; NO, nitric oxide; eNOS, endothelial nitric oxide synthase; mTOR, mammalian target of rapamycin; NLRP3, (nucleotide oligomerization domain (NOD), leucine-rich repeat (LRR) and pyrin domain (PYD)); OXPHOS, oxidative phosphorylation system; PDX-1, pancreatic and duodenal homeobox 1; PKC, protein kinase C; RAGEs, receptor for AGEs; ROS, reactive oxygen species; SOD, dismutase; JNK, c-Jun N-terminal kinase; T2D, type 2 diabetes; TXNIP, thioredoxin-interacting protein.