 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Takuro Numaga-Tomita | + 2497 word(s) | 2497 | 2021-05-14 11:31:40 | | | |
| 2 | Vivi Li | Meta information modification | 2497 | 2021-06-03 04:50:05 | | |
Video Upload Options
The heart flexibly changes its structure in response to changing environments and oxygen/nutrition demands of the body. Increased and decreased mechanical loading induces hypertrophy and atrophy of cardiomyocytes, respectively. In physiological conditions, these structural changes of the heart are reversible. However, chronic stresses such as hypertension or cancer cachexia cause irreversible remodeling of the heart, leading to heart failure. Accumulating evidence indicates that calcium dyshomeostasis and aberrant reactive oxygen species production cause pathological heart remodeling. Canonical transient receptor potential (TRPC) is a nonselective cation channel subfamily whose multimodal activation or modulation of channel activity play important roles in a plethora of cellular physiology.
1. Introduction
2. Canonical Transient Receptor Potential (TRPC) Channels
3. Cardiac Hypertrophy
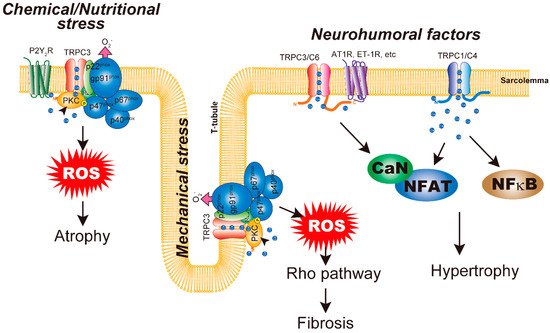
4. Cardiac Atrophy
References
- Hill, J.A.; Olson, E.N. Cardiac plasticity. New Engl. J. Med. 2008, 358, 1370–1380.
- Mihl, C.; Dassen, W.R.; Kuipers, H. Cardiac remodelling: Concentric versus eccentric hypertrophy in strength and endurance athletes. Neth. Heart J. 2008, 16, 129–133.
- Bildyug, N. Extracellular Matrix in Regulation of Contractile System in Cardiomyocytes. Int. J. Mol. Sci. 2019, 20, 5054.
- Montell, C.; Rubin, G.M. Molecular characterization of the Drosophila trp locus: A putative integral membrane protein required for phototransduction. Neuron 1989, 2, 1313–1323.
- Ramsey, I.S.; Delling, M.; Clapham, D.E. An introduction to TRP channels. Annu. Rev. Physiol. 2006, 68, 619–647.
- Vazquez, G.; Wedel, B.J.; Aziz, O.; Trebak, M.; Putney, J.W., Jr. The mammalian TRPC cation channels. BBA 2004, 1742, 21–36.
- Wes, P.D.; Chevesich, J.; Jeromin, A.; Rosenberg, C.; Stetten, G.; Montell, C. TRPC1, a human homolog of a Drosophila store-operated channel. Proc. Natl. Acad. Sci. USA 1995, 92, 9652–9656.
- Zitt, C.; Zobel, A.; Obukhov, A.G.; Harteneck, C.; Kalkbrenner, F.; Luckhoff, A.; Schultz, G. Cloning and functional expression of a human Ca2+-permeable cation channel activated by calcium store depletion. Neuron 1996, 16, 1189–1196.
- Zhu, X.; Jiang, M.; Peyton, M.; Boulay, G.; Hurst, R.; Stefani, E.; Birnbaumer, L. trp, a novel mammalian gene family essential for agonist-activated capacitative Ca2+ entry. Cell 1996, 85, 661–671.
- Liu, X.; Cheng, K.T.; Bandyopadhyay, B.C.; Pani, B.; Dietrich, A.; Paria, B.C.; Swaim, W.D.; Beech, D.; Yildrim, E.; Singh, B.B.; et al. Attenuation of store-operated Ca2+ current impairs salivary gland fluid secretion in TRPC1(-/-) mice. Proc. Natl. Acad. Sci. USA 2007, 104, 17542–17547.
- Mori, Y.; Wakamori, M.; Miyakawa, T.; Hermosura, M.; Hara, Y.; Nishida, M.; Hirose, K.; Mizushima, A.; Kurosaki, M.; Mori, E.; et al. Transient receptor potential 1 regulates capacitative Ca(2+) entry and Ca(2+) release from endoplasmic reticulum in B lymphocytes. J. Exp. Med. 2002, 195, 673–681.
- Maroto, R.; Raso, A.; Wood, T.G.; Kurosky, A.; Martinac, B.; Hamill, O.P. TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nat. Cell Biol. 2005, 7, 179–185.
- Nishida, M.; Hara, Y.; Yoshida, T.; Inoue, R.; Mori, Y. TRP channels: Molecular diversity and physiological function. Microcirculation 2006, 13, 535–550.
- Cioffi, D.L. Redox regulation of endothelial canonical transient receptor potential channels. Antioxid. Redox Signal. 2011, 15, 1567–1582.
- Yamaguchi, Y.; Iribe, G.; Nishida, M.; Naruse, K. Role of TRPC3 and TRPC6 channels in the myocardial response to stretch: Linking physiology and pathophysiology. Prog. Biophys. Mol. Biol. 2017, 130, 264–272.
- Veit, F.; Pak, O.; Brandes, R.P.; Weissmann, N. Hypoxia-dependent reactive oxygen species signaling in the pulmonary circulation: Focus on ion channels. Antioxid. Redox Signal. 2015, 22, 537–552.
- Kozai, D.; Ogawa, N.; Mori, Y. Redox regulation of transient receptor potential channels. Antioxid. Redox Signal. 2014, 21, 971–986.
- Ding, Y.; Winters, A.; Ding, M.; Graham, S.; Akopova, I.; Muallem, S.; Wang, Y.; Hong, J.H.; Gryczynski, Z.; Yang, S.H.; et al. Reactive oxygen species-mediated TRPC6 protein activation in vascular myocytes, a mechanism for vasoconstrictor-regulated vascular tone. J. Biol. Chem. 2011, 286, 31799–31809.
- Poteser, M.; Graziani, A.; Rosker, C.; Eder, P.; Derler, I.; Kahr, H.; Zhu, M.X.; Romanin, C.; Groschner, K. TRPC3 and TRPC4 associate to form a redox-sensitive cation channel. Evidence for expression of native TRPC3-TRPC4 heteromeric channels in endothelial cells. J. Biol. Chem. 2006, 281, 13588–13595.
- Spassova, M.A.; Hewavitharana, T.; Xu, W.; Soboloff, J.; Gill, D.L. A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc. Natl. Acad. Sci. USA 2006, 103, 16586–16591.
- Gottlieb, P.; Folgering, J.; Maroto, R.; Raso, A.; Wood, T.G.; Kurosky, A.; Bowman, C.; Bichet, D.; Patel, A.; Sachs, F.; et al. Revisiting TRPC1 and TRPC6 mechanosensitivity. Pflug. Archiv.: Eur. J. Physiol. 2008, 455, 1097–1103.
- Mederos y Schnitzler, M.; Storch, U.; Meibers, S.; Nurwakagari, P.; Breit, A.; Essin, K.; Gollasch, M.; Gudermann, T. Gq-coupled receptors as mechanosensors mediating myogenic vasoconstriction. EMBO J. 2008, 27, 3092–3103.
- Seth, M.; Zhang, Z.S.; Mao, L.; Graham, V.; Burch, J.; Stiber, J.; Tsiokas, L.; Winn, M.; Abramowitz, J.; Rockman, H.A.; et al. TRPC1 channels are critical for hypertrophic signaling in the heart. Circ. Res. 2009, 105, 1023–1030.
- Inoue, R.; Jensen, L.J.; Jian, Z.; Shi, J.; Hai, L.; Lurie, A.I.; Henriksen, F.H.; Salomonsson, M.; Morita, H.; Kawarabayashi, Y.; et al. Synergistic activation of vascular TRPC6 channel by receptor and mechanical stimulation via phospholipase C/diacylglycerol and phospholipase A2/omega-hydroxylase/20-HETE pathways. Circ. Res. 2009, 104, 1399–1409.
- Numaga-Tomita, T.; Oda, S.; Shimauchi, T.; Nishimura, A.; Mangmool, S.; Nishida, M. TRPC3 Channels in Cardiac Fibrosis. Front. Cardiovasc. Med. 2017, 4, 56.
- Trebak, M.; Vazquez, G.; Bird, G.S.; Putney, J.W., Jr. The TRPC3/6/7 subfamily of cation channels. Cell Calcium 2003, 33, 451–461.
- Hofmann, T.; Obukhov, A.G.; Schaefer, M.; Harteneck, C.; Gudermann, T.; Schultz, G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999, 397, 259–263.
- Storch, U.; Forst, A.L.; Pardatscher, F.; Erdogmus, S.; Philipp, M.; Gregoritza, M.; Mederos, Y.S.M.; Gudermann, T. Dynamic NHERF interaction with TRPC4/5 proteins is required for channel gating by diacylglycerol. Proc. Natl. Acad. Sci. USA 2017, 114, E37–E46.
- Vinayagam, D.; Mager, T.; Apelbaum, A.; Bothe, A.; Merino, F.; Hofnagel, O.; Gatsogiannis, C.; Raunser, S. Electron cryo-microscopy structure of the canonical TRPC4 ion channel. eLife 2018, 7.
- Tang, Q.; Guo, W.; Zheng, L.; Wu, J.X.; Liu, M.; Zhou, X.; Zhang, X.; Chen, L. Structure of the receptor-activated human TRPC6 and TRPC3 ion channels. Cell Res. 2018, 28, 746–755.
- Fan, C.; Choi, W.; Sun, W.; Du, J.; Lu, W. Structure of the human lipid-gated cation channel TRPC3. eLife 2018, 7.
- Duan, J.; Li, J.; Zeng, B.; Chen, G.L.; Peng, X.; Zhang, Y.; Wang, J.; Clapham, D.E.; Li, Z.; Zhang, J. Structure of the mouse TRPC4 ion channel. Nat. Commun. 2018, 9, 3102.
- Lichtenegger, M.; Tiapko, O.; Svobodova, B.; Stockner, T.; Glasnov, T.N.; Schreibmayer, W.; Platzer, D.; de la Cruz, G.G.; Krenn, S.; Schober, R.; et al. An optically controlled probe identifies lipid-gating fenestrations within the TRPC3 channel. Nat. Chem. Biol. 2018, 14, 396–404.
- Mederos, Y.S.M.; Gudermann, T.; Storch, U. Emerging Roles of Diacylglycerol-Sensitive TRPC4/5 Channels. Cells 2018, 7, 218.
- Bernardo, B.C.; Weeks, K.L.; Pretorius, L.; McMullen, J.R. Molecular distinction between physiological and pathological cardiac hypertrophy: Experimental findings and therapeutic strategies. Pharmacol. Ther. 2010, 128, 191–227.
- Hudlicka, O.; Brown, M.; Egginton, S. Angiogenesis in skeletal and cardiac muscle. Physiol. Rev. 1992, 72, 369–417.
- Shiojima, I.; Sato, K.; Izumiya, Y.; Schiekofer, S.; Ito, M.; Liao, R.; Colucci, W.S.; Walsh, K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J. Clin. Investig. 2005, 115, 2108–2118.
- Abel, E.D.; Doenst, T. Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc. Res. 2011, 90, 234–242.
- Taegtmeyer, H.; Sen, S.; Vela, D. Return to the fetal gene program: A suggested metabolic link to gene expression in the heart. Ann. N. Y. Acad. Sci. 2010, 1188, 191–198.
- Troncoso, R.; Ibarra, C.; Vicencio, J.M.; Jaimovich, E.; Lavandero, S. New insights into IGF-1 signaling in the heart. Trends Endocrinol. Metab.: TEM 2014, 25, 128–137.
- Dirkx, E.; da Costa Martins, P.A.; De Windt, L.J. Regulation of fetal gene expression in heart failure. BBA 2013, 1832, 2414–2424.
- Eder, P. Cardiac Remodeling and Disease: SOCE and TRPC Signaling in Cardiac Pathology. Adv. Exp. Med. Biol. 2017, 993, 505–521.
- Houser, S.R.; Molkentin, J.D. Does contractile Ca2+ control calcineurin-NFAT signaling and pathological hypertrophy in cardiac myocytes? Sci. Signal. 2008, 1, pe31.
- Sadoshima, J.; Izumo, S. The cellular and molecular response of cardiac myocytes to mechanical stress. Annu. Rev. Physiol. 1997, 59, 551–571.
- Zou, Y.; Akazawa, H.; Qin, Y.; Sano, M.; Takano, H.; Minamino, T.; Makita, N.; Iwanaga, K.; Zhu, W.; Kudoh, S.; et al. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat. Cell Biol. 2004, 6, 499–506.
- Sabourin, J.; Boet, A.; Rucker-Martin, C.; Lambert, M.; Gomez, A.M.; Benitah, J.P.; Perros, F.; Humbert, M.; Antigny, F. Ca(2+) handling remodeling and STIM1L/Orai1/TRPC1/TRPC4 upregulation in monocrotaline-induced right ventricular hypertrophy. J. Mol. Cell. Cardiol. 2018, 118, 208–224.
- Ohba, T.; Watanabe, H.; Murakami, M.; Takahashi, Y.; Iino, K.; Kuromitsu, S.; Mori, Y.; Ono, K.; Iijima, T.; Ito, H. Upregulation of TRPC1 in the development of cardiac hypertrophy. J. Mol. Cell. Cardiol. 2007, 42, 498–507.
- Chen, M.S.; Xiao, J.H.; Wang, Y.; Xu, B.M.; Gao, L.; Wang, J.L. Upregulation of TRPC1 contributes to contractile function in isoproterenol-induced hypertrophic myocardium of rat. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2013, 32, 951–959.
- Kiso, H.; Ohba, T.; Iino, K.; Sato, K.; Terata, Y.; Murakami, M.; Ono, K.; Watanabe, H.; Ito, H. Sildenafil prevents the up-regulation of transient receptor potential canonical channels in the development of cardiomyocyte hypertrophy. Biochem. Biophys. Res. Commun. 2013, 436, 514–518.
- Tang, L.; Yao, F.; Wang, H.; Wang, X.; Shen, J.; Dai, B.; Wu, H.; Zhou, D.; Guo, F.; Wang, J.; et al. Inhibition of TRPC1 prevents cardiac hypertrophy via NF-kappaB signaling pathway in human pluripotent stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 2019, 126, 143–154.
- Wu, X.; Eder, P.; Chang, B.; Molkentin, J.D. TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 7000–7005.
- Cooley, N.; Grubb, D.R.; Luo, J.; Woodcock, E.A. The phosphatidylinositol(4,5)bisphosphate-binding sequence of transient receptor potential channel canonical 4alpha is critical for its contribution to cardiomyocyte hypertrophy. Mol. Pharmacol. 2014, 86, 399–405.
- Satoh, E.; Ono, K.; Xu, F.; Iijima, T. Cloning and functional expression of a novel splice variant of rat TRPC4. Circ. J. Off. J. Jpn. Circ. Soc. 2002, 66, 954–958.
- Kirschmer, N.; Bandleon, S.; von Ehrlich-Treuenstatt, V.; Hartmann, S.; Schaaf, A.; Lamprecht, A.K.; Miranda-Laferte, E.; Langsenlehner, T.; Ritter, O.; Eder, P. TRPC4alpha and TRPC4beta Similarly Affect Neonatal Cardiomyocyte Survival during Chronic GPCR Stimulation. PLoS ONE 2016, 11, e0168446.
- Camacho Londono, J.E.; Tian, Q.; Hammer, K.; Schroder, L.; Camacho Londono, J.; Reil, J.C.; He, T.; Oberhofer, M.; Mannebach, S.; Mathar, I.; et al. A background Ca2+ entry pathway mediated by TRPC1/TRPC4 is critical for development of pathological cardiac remodelling. Eur. Heart J. 2015, 36, 2257–2266.
- Baskin, K.K.; Taegtmeyer, H. Taking pressure off the heart: The ins and outs of atrophic remodelling. Cardiovasc. Res. 2011, 90, 243–250.
- Razeghi, P.; Volpini, K.C.; Wang, M.E.; Youker, K.A.; Stepkowski, S.; Taegtmeyer, H. Mechanical unloading of the heart activates the calpain system. J. Mol. Cell. Cardiol. 2007, 42, 449–452.
- Baskin, K.K.; Rodriguez, M.R.; Kansara, S.; Chen, W.; Carranza, S.; Frazier, O.H.; Glass, D.J.; Taegtmeyer, H. MAFbx/Atrogin-1 is required for atrophic remodeling of the unloaded heart. J. Mol. Cell. Cardiol. 2014, 72, 168–176.
- Letavernier, E.; Zafrani, L.; Perez, J.; Letavernier, B.; Haymann, J.P.; Baud, L. The role of calpains in myocardial remodelling and heart failure. Cardiovasc. Res. 2012, 96, 38–45.
- Ritter, M.; Su, Z.; Xu, S.; Shelby, J.; Barry, W.H. Cardiac unloading alters contractility and calcium homeostasis in ventricular myocytes. J. Mol. Cell. Cardiol. 2000, 32, 577–584.
- Korecky, B.; Ganguly, P.K.; Elimban, V.; Dhalla, N.S. Muscle mechanics and Ca2+ transport in atrophic heart transplants in rat. Am. J. Physiol. 1986, 251, H941–H948.
- Burridge, P.W.; Li, Y.F.; Matsa, E.; Wu, H.; Ong, S.G.; Sharma, A.; Holmstrom, A.; Chang, A.C.; Coronado, M.J.; Ebert, A.D.; et al. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat. Med. 2016, 22, 547–556.
- Felker, G.M.; Thompson, R.E.; Hare, J.M.; Hruban, R.H.; Clemetson, D.E.; Howard, D.L.; Baughman, K.L.; Kasper, E.K. Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. New Engl. J. Med. 2000, 342, 1077–1084.
- Cove-Smith, L.; Woodhouse, N.; Hargreaves, A.; Kirk, J.; Smith, S.; Price, S.A.; Galvin, M.; Betts, C.J.; Brocklehurst, S.; Backen, A.; et al. An integrated characterization of serological, pathological, and functional events in doxorubicin-induced cardiotoxicity. Toxicol. Sci.: An Off. J. Soc. Toxicol. 2014, 140, 3–15.
- Zhu, W.; Soonpaa, M.H.; Chen, H.; Shen, W.; Payne, R.M.; Liechty, E.A.; Caldwell, R.L.; Shou, W.; Field, L.J. Acute doxorubicin cardiotoxicity is associated with p53-induced inhibition of the mammalian target of rapamycin pathway. Circulation 2009, 119, 99–106.
- Zhao, Y.; McLaughlin, D.; Robinson, E.; Harvey, A.P.; Hookham, M.B.; Shah, A.M.; McDermott, B.J.; Grieve, D.J. Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with Doxorubicin chemotherapy. Cancer Res. 2010, 70, 9287–9297.
- Li, J.Z.; Yu, S.Y.; Wu, J.H.; Shao, Q.R.; Dong, X.M. Paeoniflorin protects myocardial cell from doxorubicin-induced apoptosis through inhibition of NADPH oxidase. Can. J. Physiol. Pharmacol. 2012, 90, 1569–1575.
- Shimauchi, T.; Numaga-Tomita, T.; Ito, T.; Nishimura, A.; Matsukane, R.; Oda, S.; Hoka, S.; Ide, T.; Koitabashi, N.; Uchida, K.; et al. TRPC3-Nox2 complex mediates doxorubicin-induced myocardial atrophy. JCI Insight 2017, 2.
- Sudi, S.B.; Tanaka, T.; Oda, S.; Nishiyama, K.; Nishimura, A.; Sunggip, C.; Mangmool, S.; Numaga-Tomita, T.; Nishida, M. TRPC3-Nox2 axis mediates nutritional deficiency-induced cardiomyocyte atrophy. Sci. Rep. 2019, 9, 9785.
- Faigle, M.; Seessle, J.; Zug, S.; El Kasmi, K.C.; Eltzschig, H.K. ATP release from vascular endothelia occurs across Cx43 hemichannels and is attenuated during hypoxia. PLoS ONE 2008, 3, e2801.
- Paul, P.K.; Bhatnagar, S.; Mishra, V.; Srivastava, S.; Darnay, B.G.; Choi, Y.; Kumar, A. The E3 ubiquitin ligase TRAF6 intercedes in starvation-induced skeletal muscle atrophy through multiple mechanisms. Mol. Cell. Biol. 2012, 32, 1248–1259.
- Dolmatova, E.; Spagnol, G.; Boassa, D.; Baum, J.R.; Keith, K.; Ambrosi, C.; Kontaridis, M.I.; Sorgen, P.L.; Sosinsky, G.E.; Duffy, H.S. Cardiomyocyte ATP release through pannexin 1 aids in early fibroblast activation. American journal of physiology. Heart Circ. Physiol. 2012, 303, H1208–H1218.

