The
trp gene was first discovered in a phototransduction mutant of
Drosophila [
4]. A spontaneous mutation in
trp resulted in a transient receptor potential in response to continuous light. There are 28 mammalian TRP homologues, which can be further divided into six subfamilies based on genetic and functional similarities: TRPC (canonical), TRPV (vanilloid), TRPM (melastatin), TRPP (polycystin), TRPML (mucolipin), and TRPA (ankyrin). TRP channels have common structural features, including six transmembrane domains, a pore domain between the fifth and sixth transmembrane domains, and a preserved 25-amino-acid sequence termed the ‘TRP domain’. TRPC family proteins, comprising seven mammalian homologues (TRPC1–TRPC7), are the most evolutionally preserved TRP channels with regard to the original form identified in
Drosophila. Therefore, it was believed and later demonstrated that they function as receptor-activated calcium channels [
5]. Among them, TRPC4 and TRPC5 have about 65% amino acid homology, while TRPC3, TRPC6, and TRPC7 have about 75% homology [
6]. TRPC1 was originally reported as a candidate subunit of store-operated Ca
2+ channels [
7,
8,
9,
10]. The functional importance of TRPC1 channels is manifested by the promotion of functional coupling between endoplasmic reticulum and the plasma membrane during receptor-induced Ca
2+ signaling [
11]. In addition, it has been shown that mechanical stretch activates TRPC1 in mammalian cells [
12]. Thus, TRPC proteins have two important roles: one as a functional channel activated by mechanical stretch or endoplasmic reticulum store depletion and the other as a platform to organize receptor-activated Ca
2+ channels and intracellular signaling molecules for efficient signal transduction [
13].
TRPC channels are widely recognized to be activated downstream of phospholipase C (PLC)-coupled receptors such as G protein-coupled receptors (GPCRs) and receptor tyrosine kinases [
13]. As a result of their universal activation mechanism in many cell types, TRPC channels play important roles in basic cellular responses including proliferation, differentiation, and death in response to various environmental stimuli. Recent findings indicate that in addition to PLC-mediated activation, TRPC channels are multimodally activated by environmental stimuli such as mechanical stretch, hypoxia, and oxidative stress [
14,
15,
16,
17,
18,
19]. In particular, TRPC1 and TRPC6 are suggested to be activated by mechanical stimuli [
12,
20]. Later studies suggested that mechanical activation of TRPC1 and TRPC6 is mediated by phospholipase-dependent mediators [
21,
22,
23,
24]. Physiological or pathophysiological conditions expose the heart to changes in mechanical loading and oxygen supply, as well as oxidative stress, especially in the pathological heart. Therefore, TRPC channels play important roles in transducing these environmental stimuli into Ca
2+ and/or chemical signals within cardiomyocytes.
All TRPC channels are expressed in cardiomyocytes, except TRPC2. The expression of some of these channels is not high in normal conditions but is increased in the failing heart [
25]. According to studies using heterologous expression systems, the TRPC family can be further subdivided into two groups: TRPC1/C4/C5 and TRPC3/C6/C7. Apart from several observations, TRPC channels form functional channels by forming homo- or heterotetramers within these groups. So far, heterologous expression systems have demonstrated that the difference between these two groups involves activation by the PLC product of diacylglycerol (DAG) [
26]. TRPC3/C6/C7 is activated directly by DAG, whereas TRPC1/C4/C5 is not [
27]. However, Storch et al. recently revealed that DAG activates TRPC4/TRPC5 [
28]. Structural analysis with cryogenic electron microscopy revealed the structural conservation of TRPC family channels [
29,
30,
31,
32]. This is further supported by the fact that a glycine residue in the selective filter of TRPC3 is both critical to the recognition of lipid mediators and conserved among TRPC channels [
33]. Although the actual mechanism of TRPC channel downstream surface receptor activation is still not completely understood [
34], its importance in cardiac plasticity has been extensively documented in the model of neurohumoral-factor-induced cardiac remodeling. The original concept of TRPC channels in cardiac remodeling is quite simple. First, major pro-hypertrophic factors such as angiotensin II (AngII) and endothelin-1 activate their receptors, which couple to PLC and downstream TRPC channels. Second, TRPC channels mediate Ca
2+ influx, which activates major hypertrophic signaling of calcineurin (CaN)/nuclear factor of activated T cells (NFAT). However, recent findings indicate that the involvement of TRPC channels is more complex and chaotic. At present, it is not easy to understand and conceptualize the importance of TRPC channels in cardiac plasticity. Therefore, we herein review recent findings about TRPC channels in cardiac remodeling and raise models and questions that need to be addressed in the near future.
3. Cardiac Hypertrophy
The heart changes its size and contractility in response to increased or decreased hemodynamic load caused by body demand for oxygen and nutrition supplies [
1,
2]. The best-known adaptive change of the heart is hypertrophy. However, continued increase of the pressure for hypertrophy leads to maladaptive changes of the heart, ultimately resulting in heart failure. Numerous signaling pathways have been implicated in cardiac hypertrophy [
71]. Cardiac hypertrophy is an inherently adaptive response. Athletes or pregnant women experience cardiac hypertrophy, but most of this is completely reversible. The heart size returns to a normal level by attenuation of physical activity or after delivery. Several studies have demonstrated the difference between physiological and pathological cardiac hypertrophy [
71]. Physiological and pathological hypertrophy have distinct features at structural, metabolic, and molecular levels. Physiological hypertrophy is accompanied by angiogenesis. In contrast, pathological hypertrophy is accompanied by interstitial fibrosis and reduced capillary densities, which result in myocardial ischemia and lead to heart failure [
72,
73]. During cardiac hypertrophy, the heart remodels its metabolic pathways to increase ATP production. In physiological hypertrophy, fatty acid and glucose oxidation are increased, in contrast to decreases observed during pathological hypertrophy [
74]. In response, the glycolytic pathway is upregulated in pathological hypertrophy, likely to compensate for reduced mitochondrial ATP production. Interestingly, the fetal heart depends on the glycolytic pathway because of the poor availability of fatty acids [
71]. Moreover, it has been well recognized that the molecular features of pathological hypertrophy include increased fetal gene expression [
75]. Thus, pathological hypertrophy occurs concurrently with a regression to a fetal heart phenotype with regard to metabolic and genetic aspects.
The best-known signaling pathway regulating physiological hypertrophy is the insulin-like growth factor 1-PI3K(p110α)-Akt pathway. Insulin-like growth factor 1 is important for postnatal development and in the exercised adult heart [
76]. Two major pathways have been implicated in pathological cardiac hypertrophy; one is neurohumoral-factor-dependent while the other is biomechanical-stress-dependent [
1]. The neurohumoral-factor-dependent pathway is mediated by Gq-type GPCRs, such as AngII receptor type 1 (AT-1) and endothelin receptor. Many signaling pathways participate in pathological hypertrophy, such as small G proteins, mitogen-activated protein kinases, histone deacetylases, and calcium signaling, which are integrated under transcriptional control by several transcription factors. [
71,
77]. Altered Ca
2+ signaling, one trigger for cardiac hypertrophy, is likely attributable to changes of PLC and phosphatidylinositol-turnover-dependent increases in intracellular Ca
2+ [
53,
71,
78]. Although how cardiomyocytes sense and transduce biomechanical signals into hypertrophy is still largely unknown, some reports have demonstrated increased autocrine and paracrine secretion of pro-hypertrophic neurohumoral factors and growth factors in mechanically stressed cardiomyocytes [
79]. Alternatively, ligand-independent activation of AT-1 receptors by mechanical stretch might mediate abnormal Ca
2+ increases [
80]. Several Ca
2+ signaling molecules have been identified as critical regulators of cardiac hypertrophy. Most notably, activity of the Ca
2+-dependent transcriptional pathway of CaN/NFAT is important for pathological cardiac hypertrophy [
78]. Therefore, it is very likely that Ca
2+-permeable channels play important roles in abnormal Ca
2+ signaling in pathological cardiac hypertrophy. As a candidate molecular entity for those Ca
2+ channels, TRPC channels have been intensively studied.
Both mRNA and protein expression levels of TRPC1 are increased in the hearts of cardiac hypertrophy model rats [
55,
56,
81]. In addition, several experiments demonstrated that TRPC1 plays a critical role in neurohumoral-factor-induced cardiac hypertrophy in vitro [
81,
82]. TRPC1-knockout mice showed no cardiac hypertrophy after transverse aortic constriction [
23]. In addition to these animal model studies, patients with cardiac hypertrophy or heart failure showed increased expression of TRPC1 mRNA [
83]. In addition, the involvement of TRPC1 in cardiac hypertrophy was demonstrated in human embryonic stem cell derived cardiomyocytes [
83]. As hypertrophied cardiomyocytes have larger SOCE, upregulation of TRPC1 contributes to increased responses that subsequently activate the CaN/NFAT pathway, leading to pathological hypertrophy [
55,
56]. In addition to this well-known pathway, RNA sequencing of human cardiomyocytes indicates that downstream signaling of TRPC1 involves the NF-κB pathway [
83].
Suppression of TRPC4 activity by expression of a dominant-negative mutant of TRPC4 suppressed transverse aortic constriction induced cardiac hypertrophy [
36]. Furthermore, the heart expresses several TRPC4 splice variants from α to γ [
84,
85]. Among these variants, only TRPC4a can interact with PLCβ1b and is expressed on the sarcolemma. Overexpression of TRPC4a in cardiomyocytes induces cardiomyocyte hypertrophy [
84]. However, chronic treatment with pro-hypertrophic AngII does not further increase the size of cardiomyocytes. In addition, TRPC4b overexpression has nothing to do with resting or AngII-induced cardiac hypertrophy. In both TRPC4a- and TRPC4b-overexpressing cardiomyocytes, basal activity of the CaN/NFAT pathway is increased [
86]. Consistent with these findings, the most important role of TRPC1/C4 seems to be the pathway for background Ca
2+ entry in cardiomyocytes to fine-tune systolic and diastolic intracellular Ca
2+ concentration [
87]. These results suggest that TRPC4 channel isoforms have different functions in cardiomyocytes and affect downstream targets of universal CaN/NFAT signaling in cardiac hypertrophy ().
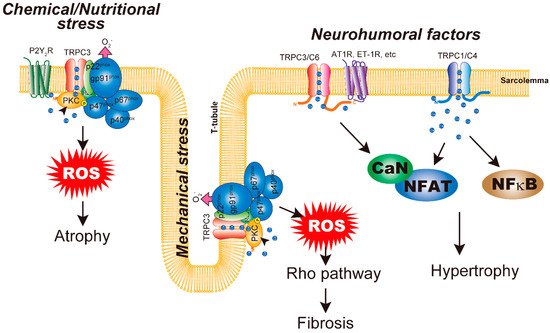
Figure 1. Schematic representation of canonical transient receptor potential (TRPC) channels in cardiac plasticity. Neurohumoral-factor-induced cardiac hypertrophy is mediated by the CaN/NFAT pathway. TRPC3/C6 channels contribute to Ca2+ increase downstream of GPCR activation. TRPC1/C4 channels function to regulate background Ca2+ entry and the basal level of CaN/NFAT activation in cardiomyocytes. Homomeric TRPC3 channels are upregulated by mechanical stresses. TRPC3 prevents Nox2 from undergoing proteasomal degradation, leading to increased Nox2 protein abundance. TRPC3 is also important for enzymatic activation of Nox2. TRPC3–Nox2 coupling mediates chemical (DOX) and nutritional stress-induced cardiac atrophy. Chemical/nutritional stresses evoke ATP release from cardiomyocytes, which activates Gq-coupled P2Y2 receptors.
4. Cardiac Atrophy
Cardiac muscle mass is decreased in response to a reduction of hemodynamic load, which is known to be caused by microgravity, long-term bed rest, or cancer cachexia. Cardiac atrophy is mediated by increased protein breakdown by the ubiquitin–proteasome system, Ca
2+-dependent calpain system, lysosomal system, and autophagy [
101]. Hemodynamic unloading by heart transplantation has revealed that unloading stresses increase the expression of calpain proteases [
102] and E3 ubiquitin ligases MuRF1 and atrogin-1 [
103]. Interestingly, Taegtmeyer’s group observed similar induction of a fetal gene program in atrophied hearts as demonstrated in hypertrophied hearts [
75,
101]. Activation of the fetal gene program seems to be an adaptive response to metabolic changes of the heart. Therefore, atrophic responses of the heart are not a passive reduction of muscle mass but an active remodeling of heart structure and function occurring in response to a change of hemodynamic load. Compared with hypertrophic responses, underlying mechanisms of cardiac atrophy remain largely elusive. Ca
2+ signaling likely plays important roles in cardiac atrophy, as the calpain system requires increases of intracellular Ca
2+ [
104], and some reports have demonstrated Ca
2+ dyshomeostasis in unloaded cardiomyocytes [
105,
106]. The molecular mechanism underlying cardiac atrophy is presumed based on observations of skeletal muscle atrophy. In skeletal muscle, mechanical unloading causes mitochondrial dysfunction and reactive oxygen species (ROS) production.
In contrast to cardiac hypertrophy, the molecular mechanism of cardiac atrophy remains largely unknown. Doxorubicin (DOX) is a highly effective anticancer drug prescribed for a wide range of cancers. However, the dose-dependent cardiotoxic effects of DOX limits its long-term use. Specifically, the cardiotoxic effects of DOX include arrhythmias induced by its acute effect, the development of left ventricular systolic dysfunction (which leads to dilated cardiomyopathy), and congestive heart failure induced by its chronic effect [
107,
108]. It was demonstrated that DOX initially induces cardiac shrinkage followed by the induction of myocardial apoptosis and interstitial fibrosis at later stages of left ventricular dilated cardiomyopathy [
109,
110]. In
Nox2–/– mice, DOX-induced structural remodeling was completely attenuated [
111]. DOX treatment has been shown to increase Nox2 expression in the heart [
111,
112,
113]. Strikingly, the reduction of Nox2 expression by treatment with paeoniflorin suppressed DOX-induced cardiomyocyte apoptosis [
112]. In our mouse model, DOX treatment induced cardiac atrophy from 2 weeks after treatment. DOX treatment increased Nox2 protein abundance, which was significantly suppressed by the absence of TRPC3. TRPC3-knockout mouse hearts maintained both whole-heart and cross-sectional areas of cardiomyocytes [
113]. Therefore, similar to mechanical overloading, TRPC3 can couple with Nox2 in DOX-induced cardiac atrophy. We recently revealed that nutritional deficiency caused by glucose and amino acid starvation and hypoxia induces cardiomyocyte atrophy [
114]. Nutritional stresses evoke intracellular ATP release to the extracellular space, as has been demonstrated in cardiomyocytes and other cells [
115,
116,
117]. Extracellular ATP is the actual signal inducing atrophic responses in cardiomyocytes. As mentioned above, it is known that extracellular ATP does not induce cardiac hypoxia, even with a significant increase of the CaN/NFAT pathway. Atrophic responses of cardiomyocytes could be observed with stimulation of around 1 mM extracellular ATP. A submaximal concentration of ATP (e.g., 100 μM) does not induce atrophic responses. However, the dose responses of ATP to cardiomyocyte atrophy correlated well with intracellular ROS production. Indeed, high ATP stimulation does not increase TRPC3 or Nox2 protein abundances but enhances the interaction between them. Atrophic responses caused by both nutritional stresses and high extracellular ATP are sensitive to the specific P2Y
2 receptor antagonist, indicating that the P2Y
2 receptor is upstream of TRPC3–Nox2 coupling. Interestingly, DOX-induced atrophic responses were also suppressed by a P2Y
2 receptor antagonist. These results suggest the critical involvement of intracellular ATP release and subsequent TRPC3 activation in stress-sensing and transduction of cardiomyocytes ().