 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Oleg Timofeev | + 2861 word(s) | 2861 | 2021-05-26 04:11:29 | | | |
| 2 | Nora Tang | Meta information modification | 2861 | 2021-05-31 03:07:37 | | |
Video Upload Options
p53 is a tumor suppressor that is mutated in half of all cancers. The high clinical relevance has made p53 a model transcription factor for delineating general mechanisms of transcriptional regulation. p53 forms tetramers that bind DNA in a highly cooperative manner. The DNA binding cooperativity of p53 has been studied by structural and molecular biologists as well as clinical oncologists. These experiments have revealed the structural basis for cooperative DNA binding and its impact on sequence specificity and target gene spectrum. Cooperativity was found to be critical for the control of p53-mediated cell fate decisions and tumor suppression. Importantly, an estimated number of 34,000 cancer patients per year world-wide have mutations of the amino acids mediating cooperativity, and knock-in mouse models have confirmed such mutations to be tumorigenic. While p53 cancer mutations are classically subdivided into “contact” and “structural” mutations, “cooperativity” mutations form a mechanistically distinct third class that affect the quaternary structure but leave DNA contacting residues and the three-dimensional folding of the DNA-binding domain intact. In this review we discuss the concept of DNA binding cooperativity and highlight the unique nature of cooperativity mutations and their clinical implications for cancer therapy.
1. Introduction
p53 is a transcription factor that has its evolutionary origin in unicellular choanoflagellates and early metazoans [1]. Its functions in protection of genome integrity in somatic cells have crystallized later in vertebrate organisms, where it evolved into a tumor suppressor and deserved recognition as one of the most powerful ones [2]. The human TP53 gene can give rise to several splice variants [3]. Among them, the full-length isoform p53α is the most abundant and best characterized. It consists of 393 amino acids and can be subdivided into several functional domains (Figure 1a,b): two N-terminal transactivation domains (TADI and II, amino acids 1–43 and 43–63); a short proline-rich domain (PRD, aa 64–93), involved in protein–protein interactions and apoptosis; a core DNA-binding domain (DBD, aa 94–292) that enables sequence-specific DNA binding; a hinge region (aa 293–323) where a bipartite nuclear localization signal (NLS) is located; an oligomerization domain (OD aa 324–355) that mediates tetramerization of p53 molecules; and a carboxy-terminal regulatory domain (CTD aa 356–393), which binds DNA in a sequence non-specific manner and regulates p53 function and stability [1][4][5].
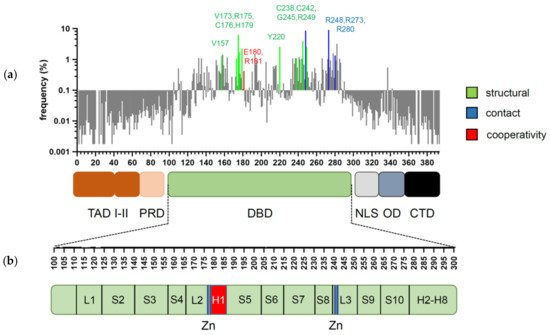
Figure 1. Domain structure of p53 and distribution of cancer-associated mutations. (a) Frequency and distribution of TP53 mutations detected in somatic tumors. Highlighted are the codons most frequently affected by structural (green), contact (blue), and cooperativity (red) mutations (references in text). (b) Secondary structure of p53 DBD. Residues responsible for zinc (Zn) ion coordination (blue) and H1 helix (red) are highlighted.
p53 undergoes complex post-translational modifications that determine its stability, cellular localization, and transcriptional activity [6]. Upon stress, specific phosphorylation and acetylation patterns of the TAD and CTD are set and lead to stabilization and activation of p53 [7], which cooperatively acts as a homotetramer in sequence-specific DNA binding and transcriptional regulation. Numerous target genes trigger a range of cellular programs—from repair of cellular damage to elimination of harmed cells, depending on stress impact and damage level [8]. Besides transcriptional functions, p53 exerts transcription-independent activities in regulation of apoptosis [9], necrosis [10], autophagy [11], metabolism [12][13], and DNA replication [14] and repair [15]. Multiple p53-controlled effector mechanisms add to its power as a tumor suppressor and create strong selective pressure against p53 in tumorigenesis.
Mutations, that hit the TP53 gene are found in virtually all cancer types, but the frequency of inactivating mutations may vary from less than 5% in neuroblastoma to above 90% in ovarian and small-cell lung cancer [16][17][18][19][20]. Among all genetic alterations that affect TP53 in tumors, missense mutations prevail, the majority of which cluster in the central DBD, underscoring its significance for p53′s tumor suppressive activity [16][20] (Figure 1). Mutations that occur with an overall frequency >1% are considered as “hotspots” and together represent about 30% of all missense mutations [21][22]. The role of these hotspot mutations for p53 structure and function is well described [23][24]. Several high-throughput screens were undertaken to investigate the remaining 70% of p53 variants [25][26][27], but the precise mechanisms how they affect p53 are less well understood. With respect to transcriptional and tumor-suppressive p53 activities, the majority are loss-of-function (LOF) mutations, which, however, affect p53 to different degrees—from partial impairment to complete inactivation. Furthermore, several hotspot mutations have been shown to bestow p53 with neomorphic, oncogenic functions and are commonly referred to as gain-of-function (GOF) mutants, which promote tumor progression, metastasis, and resistance to therapy.
Since a number of recent reviews provide comprehensive information about these aspects of p53 mutations [22][28][29], we focus this review on a new class of p53 mutations that undermine the cooperative nature of DNA binding and discuss the role of these mutations in cancer development and therapy response and implications for p53 functions in tumor suppression.
2. Cooperative DNA Binding by p53
p53 can bind DNA in two modes: sequence-specifically via the DBD and in a sequence-independent manner through the CTD [30]. Like many other transcription factors, p53 forms oligomers, and the transcriptionally active state of p53 requires the assembly of homotetramers [31]. In vitro experiments suggested that p53 dimers are assembled co-translationally and are predominant under basal conditions [32][33][34], whereas stress-induced post-translational modifications promote tetramerization [35][36]. A recent in vivo study has shown that in non-stressed cells monomers and dimers composed the major p53 pool, but DNA damage triggered rapid tetramerization [37]. The ability of p53 to bind double-stranded DNA in a sequence-specific manner is determined by the DBD, which forms an immunoglobulin-like β-sandwich structure composed of a loop-sheet-helix motif and two large loops (L2 and L3) and serves as a basic scaffold for the DNA-binding surface. The loop-sheet-helix motif that includes the L1 loop binds the minor groove, whereas the L2 and L3 loops, stabilized by a zinc ion, dock to the major groove [31][38]. A typical p53 response element (RE) contains two decameric RRRCWWGYYY (R = A,G; W = A,T; Y = C,T) half-sites separated by spacers of 0–18 base pairs (bp) [39][40]. Structural studies demonstrated that wild-type p53 proteins form tetrameric complexes on DNA, known as “dimers of dimers”, where each clamp-like symmetrical dimer binds one half-site of the RE, and two dimers occupy the full RE (Figure 2) [31][41][42][43][44][45][46].
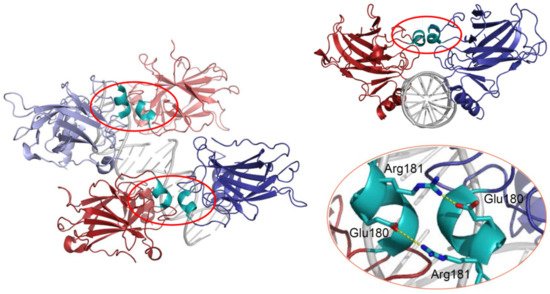
Figure 2. 3D structure of the p53 DBD tetramer in complex with DNA (based on RCSB Protein Data Bank ID:2AHI). H1 helices are highlighted in cyan; ionic bonds between negatively charged Glu180 and positively charged Arg181 are shown in yellow.
Assembly of p53 tetramers on DNA occurs in a highly cooperative manner. In general, cooperativity is a common biochemical phenomenon observed when a protein or protein complex contains multiple identical or near-identical ligand binding sites, and ligand binding to any one site increases (positive cooperativity) or decreases (negative cooperativity) the apparent affinity at the others. Cooperative binding can be mathematically described by the Hill equation, where the Hill coefficient functions as a quantitative measure of cooperativity [47]. One of the best-known examples of positive cooperativity is oxygen binding by hemoglobin, where binding of each oxygen molecule increases the binding affinity for the next until the hemoglobin tetramer is fully saturated with oxygen at all four binding sites [48]. Many other molecular assemblies exhibiting cooperative binding have been studied, including multimeric enzymes and transcription factors such as lambda phage repressor. In a very similar manner, DNA binding of p53 also displays positive cooperativity relying on the assembly of a tetrameric p53–DNA complex that is stabilized by protein–protein interactions between the four monomers [41]. Primarily, tetramerization of p53 monomers is mediated by the OD, which consists of a short β-strand and an α-helix that provide an interaction surface for dimerization. Two primary dimers associate through their α-helices and build a four-helix bundle, stabilized by hydrophobic interactions [49]. Integrity of the OD is essential for tetramerization and p53 functional activity. However, it has been shown that isolated DBDs upon interaction with p53 REs form tetramers also independent of the OD, although with 10- to 1000-fold lower binding affinity [34][50][51]. These findings indicated existence of direct protein–protein interactions between DBDs. These interactions among p53 dimers and tetramers stabilize protein/DNA complexes and shape p53′s cistrome and transcriptional activity [52][53]. Early structural models of p53 DBD/DNA complexes pointed at the H1 helix as the structural basis for cooperative interactions between core domains [31][51]. Later models based on X-ray crystallography and NMR data indicated that the DBD interaction interface is formed by residues from the H1 helix (Pro177, His178, Glu180, Arg181) and several residues from the L3 loop (Met243, Gly244) [44][54]. The H1 helix is a short α-helical structure (Pro177-Cys182) located within the L2 loop, adjacent to the DNA-binding core region but not involved in direct contact with DNA (Figure 2). Association of p53 with its RE brings the antiparallel oriented H1 helices of monomers in proximity and allows interactions that stabilize the entire complex. X-ray crystallography, NMR, and biochemical and biophysical studies pointed to an essential role of these structures for cooperative DNA binding and identified Glu180 and Arg181 as key residues that mediate reciprocal electrostatic interactions between H1 helices [42][44][54][55][56][57]. The double salt bridges formed by these oppositely charged amino acids maintain p53′s intra-dimer interactions and affect the strength of sequence-specific DNA binding [52][55][56][58]. Intriguingly, the CTD that binds DNA in a sequence-independent manner contributes to cooperativity by inducing conformational changes within the DBD that enhance sequence-specific binding [59]. The primary structure of the H1 helix is highly conserved among p53 proteins of different vertebrate species and in the p53 family members p63 and p73, but the salt bridge itself is absent in p63 and p73 [55][57][60][61].
The degree of DNA binding cooperativity is determined by structural DNA properties encoded in the RE sequence—especially in its central WW dinucleotide [62]. Even though these bases are not directly contacted by p53 residues, they are highly conserved and determine the torsional flexibility of the half-site and thus define the energy needed for DNA twisting [62]. Since DNA binding of p53 induces significant RE bending and twisting, the exact WW dinucleotide sequence strongly affects binding [50][51][57][63]. In the case of a torsionally more flexible CATG, the p53 DBD binds with low cooperativity, while REs containing more rigid CAAG, and CTAG are bound with up to 3 orders of magnitude higher cooperativity and only when present in two contiguous p53 half-sites [62]. Additionally, REs containing spacers between two half-sites are bound with high cooperativity [62]. As transactivation relies on DNA binding and DNA-protein complex stability, cooperativity is a major determinant of p53′s transactivation function.
3. Cancer-Associated Mutations of the DBD—Structural Implications
The vast majority of cancer-associated mutations in the TP53 gene are non-synonymous missense substitutions that give rise to more than 2000 mutant p53 variants [20][64]. Remarkably, over 95% of these mutations map to the core DBD [22]. The most frequent somatic “hotspot” mutations are R175H, R248Q/W, R273C/H, R282W, Y220C, G245S R249S, and V157F [20]. Based on the mechanism of action, mutations can be divided into three groups: structural, contact, and cooperativity mutations (Figure 1a).
3.1. Stuctural Mutations
In contrast to the intrinsically unfolded N- and C-terminal domains, the DBD is well structured. However, its stability is rather low: wild-type p53 denatures at 42–45 °C, and even at normal body temperature, the DBD in the context of the full-length protein has an unfolding half-life of 37 min, whereas the isolated DBD unfolds in just 9 min [65][66]. This low native stability of p53 makes it very sensitive to destabilizing mutations, which cause local or global unfolding. Many structural mutations that affect the β-sandwich region (such as V143A, V157F, Y220C, and F270C/L) or the loop-sheet-helix motif (R282W) are highly destabilizing: they reduce the thermodynamic stability of the protein by >3 kcal/mol, lowering the melting temperature by 5–7 °C [66][67][68]. As a result, the mutant protein is globally unfolded and unable to bind DNA at 37 °C but retains a wild-type-like conformation and substantial transcriptional activity at sub-physiological temperatures—so called “temperature-sensitive” mutants [69]. Because of the crucial role of the zinc ion for the core structure of the DNA-binding interface encoded by the L2-L3 loops, mutations that impact zinc binding also have a severely destabilizing effect. Non-hotspot mutations such as C176F, H179R, C238Y, and C242S directly affect zinc ligation, leading to a strongly reduced thermostability of the DBD [24][67]. The most frequent cancer mutation R175H heavily reduces zinc-binding affinity by destroying the zinc coordination sphere, which results in global unfolding at physiological and sub-physiological temperatures, making p53 completely inactive [70][71]. Other cancer-associated mutations, which hit this site with much lower frequency (such as R175C/L/P/S), seem to be less detrimental for zinc binding and have only moderate or weak effects on p53 functionality [24][72]. Mutations that affect the DNA-interacting surface can also cause local structural distortions that affect the DNA-binding proficiency of p53 to different degrees. The G245S and R249S hotspot mutations strike the L3 loop in the minor-groove-binding region. Whereas the G245S leads to small conformational changes (which, however, result in a substantial decrease in sequence-specific DNA binding), R249S has a more general impact on L3 loop conformation and DBD stability (it destabilizes the core domain by ∼2 kcal/mol), drastically affecting DNA binding [66][67][68].
3.2. Contact Mutations
Contact mutations most frequently affect the Arg248, Arg273, or Arg280 residues that are directly interacting with DNA. Arg248 is essential for docking into the minor groove and interacts with the regions flanking the core sequence of each half-site of RE [58]. Arg273 binds to the central CWWG site, providing important contacts to the DNA backbone. Arg280 anchors in the major groove, interacting with the conserved G in the CWWG sequence [42]. Arg248 and Arg273 are mutational hot-spots: R248Q/W and R273C/H substitutions represent 10–20% of all cancer-associated missense mutations detected in the DBD [20][22]. Contact mutations have only a minute effect on thermodynamic properties of the p53 protein and do not cause substantial structural perturbations [31][66][67] but drastically weaken sequence-specific DNA binding and thus disable p53′s transcriptional activity. For example, in vitro experiments with p53 tetramers containing DBD and OD showed that the R273H mutation reduced binding to the high affinity GADD45 promoter by 1000-fold [66].
3.3. Cooperativity Mutations
DBD mutations that affect the cooperative nature of p53 DNA binding are so-called “cooperativity” mutations. A number of cooperativity mutations at residues Glu180 (E180A/D/G/K/Q/V) and Arg181 (C/H/G/L/P/S) are found as somatic mutations in various types of sporadic cancer and germ-line mutations associated with the hereditary Li-Fraumeni or Li-Fraumeni-like cancer susceptibility syndromes. Together, these mutations account for 0.5–0.6% of all p53 missense mutations. Given that p53 is mutated in approximately 50% of all cancers and that 70% of these are missense mutations, this results in an estimated world-wide number of 34,000 cancer cases per year [20][22]. The distribution of cooperativity mutations across different cancer types is highly similar to all other missense mutations, showing only a slight overrepresentation of cooperativity mutations in non-small cell lung and bladder carcinoma and underrepresentation in colorectal and ovarian carcinoma (Figure 3). The most frequent cooperativity mutants (E180K, R181C, R181H) showed a selective loss of apoptosis, in parallel with reduced promoter binding and transactivation of apoptosis-related gene targets but retained substantial activity in mounting cell cycle arrest [52]. Another cooperativity mutant R181L, which is detected in somatic tumors and LFS patients, induced cell cycle arrest but failed to trigger apoptosis when ectopically expressed in p53-null cells [52][73].
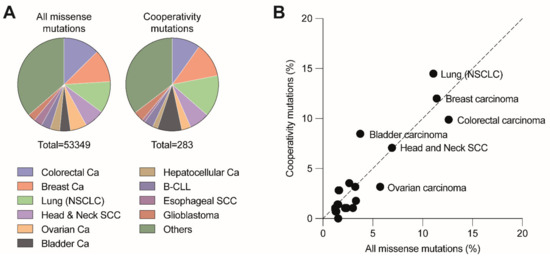
Figure 3. Distribution of cooperativity mutations across different cancer types. (A) Pie charts depict the distribution of all missense and cooperativity mutations across the listed cancer types. (B) Cancer type distribution of cooperativity mutations correlates with the distribution of all missense mutations (R2 = 0.8779, p < 0.001). Analysis based on all tumor samples with p53 missense mutations listed in the UMD TP53 Mutation Database (https://p53.fr/tp53-database, Release 2017_R2, accessed on 12 May 2021).
In addition to these naturally occurring mutations, charge-neutralizing (E180L and R181L) and, in particular, charge-inverting (E180R and R181E) mutations were employed to experimentally weaken or disrupt the H1 helix salt bridges and delineate the functional impact of altered cooperativity. Of note, engineering charge-inversion mutations requires substitution of two (E180R) or three nucleotides (R181E), respectively. However, more than 99% of p53 missense mutations in cancer patients are single-nucleotide substitutions, providing an explanation for why these charge-inversion mutations have not been observed in cancer cells so far. Overlaying the NMR solution structure of charge-inverting mutations with the wild-type DBD showed differences in chemical shifts only for signals of residues within the H1 helix or near the specifically mutated residues [55]. Residues further away were only slightly affected by these mutations or not affected at all, which affords the conclusion that salt bridge mutants are folded in the native conformation and clearly distinguishes these cooperativity mutations from the other two classes of DBD mutations. Importantly, the DNA binding deficiency of these charge-inversion mutants is entirely rescued when the salt bridges are reconstituted by combination of the two mutant proteins (E180R+R181E) or introduction of both mutations into the same p53 molecule (E180R;R181E double mutant) [52][53][55][74]. This highlights that the cellular and organismal phenotypes resulting from these mutations are solely explained by the disruption of the H1 helix salt bridges and underlines the value of engineered charge-inversion mutations for studying the functional role of DNA binding cooperation in tumor suppression and beyond.
References
- Belyi, V.A.; Ak, P.; Markert, E.; Wang, H.; Hu, W.; Puzio-Kuter, A.; Levine, A.J. The origins and evolution of the p53 family of genes. Cold Spring Harb. Perspect. Biol. 2010, 2, a001198.
- Levine, A.J. The many faces of p53: Something for everyone. J. Mol. Cell Biol. 2019, 11, 524–530.
- Khoury, M.P.; Bourdon, J.C. The isoforms of the p53 protein. Cold Spring Harb. Perspect. Biol. 2010, 2, a000927.
- Vieler, M.; Sanyal, S. P53 isoforms and their implications in cancer. Cancers 2018, 10, 288.
- Joerger, A.C.; Fersht, A.R. The tumor suppressor p53: From structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2010, 2, a000919.
- Kruse, J.P.; Gu, W. Modes of p53 Regulation. Cell 2009, 137, 609–622.
- Dai, C.; Gu, W. P53 post-translational modification: Deregulated in tumorigenesis. Trends Mol. Med. 2010, 16, 528–536.
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078.
- Marchenko, N.D.; Moll, U.M. Mitochondrial death functions of p53. Mol. Cell. Oncol. 2014, 1, e955995.
- Vaseva, A.V.; Marchenko, N.D.; Ji, K.; Tsirka, S.E.; Holzmann, S.; Moll, U.M. P53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012, 149, 1536–1548.
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687.
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. P53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316.
- Zhao, Y.; Chaiswing, L.; Velez, J.M.; Batinic-Haberle, I.; Colburn, N.H.; Oberley, T.D.; St. Clair, D.K. p53 translocation to mitochondria precedes its nuclear translocation and targets mitochondrial oxidative defense protein-manganese superoxide dismutase. Cancer Res. 2005, 65, 3745–3750.
- Hampp, S.; Kiessling, T.; Buechle, K.; Mansilla, S.F.; Thomale, J.; Rall, M.; Ahn, J.; Pospiech, H.; Gottifredi, V.; Wiesmüller, L. DNA damage tolerance pathway involving DNA polymerase ι and the tumor suppressor p53 regulates DNA replication fork progression. Proc. Natl. Acad. Sci. USA 2016, 113, E4311–E4319.
- Linke, S.P.; Sengupta, S.; Khabie, N.; Jeffries, B.A.; Buchhop, S.; Miska, S.; Henning, W.; Pedeux, R.; Wang, X.W.; Hofseth, L.J.; et al. p53 interacts with hRAD51 and hRAD54, and directly modulates homologous recombination. Cancer Res. 2003, 63, 2596–2605.
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 2019, 28, 1370–1384.
- Campbell, P.J.; Getz, G.; Korbel, J.O.; Stuart, J.M.; Jennings, J.L.; Stein, L.D.; Perry, M.D.; Nahal-Bose, H.K.; Ouellette, B.F.F.; Li, C.H.; et al. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93.
- Gerstung, M.; Jolly, C.; Leshchiner, I.; Dentro, S.C.; Gonzalez, S.; Rosebrock, D.; Mitchell, T.J.; Rubanova, Y.; Anur, P.; Yu, K.; et al. The evolutionary history of 2,658 cancers. Nature 2020, 578, 122–128.
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339.
- Leroy, B.; Anderson, M.; Soussi, T. TP53 mutations in human cancer: Database reassessment and prospects for the next decade. Hum. Mutat. 2014, 35, 672–688.
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876.
- Stiewe, T.; Haran, T.E. How mutations shape p53 interactions with the genome to promote tumorigenesis and drug resistance. Drug Resist. Updates 2018, 38, 27–43.
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713.
- Joerger, A.C.; Fersht, A.R. The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu. Rev. Biochem. 2016, 85, 375–404.
- Kato, S.; Han, S.-Y.; Liu, W.; Otsuka, K.; Shibata, H.; Kanamaru, R.; Ishioka, C. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 8424–8429.
- Kotler, E.; Shani, O.; Goldfeld, G.; Lotan-Pompan, M.; Tarcic, O.; Gershoni, A.; Hopf, T.A.; Marks, D.S.; Oren, M.; Segal, E. A Systematic p53 Mutation Library Links Differential Functional Impact to Cancer Mutation Pattern and Evolutionary Conservation. Mol. Cell 2018, 71, 178–190.
- Boettcher, S.; Miller, P.G.; Sharma, R.; McConkey, M.; Leventhal, M.; Krivtsov, A.V.; Giacomelli, A.O.; Wong, W.; Kim, J.; Chao, S.; et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 2019, 365, 599–604.
- Pavlakis, E.; Stiewe, T. p53′s extended reach: The mutant p53 secretome. Biomolecules 2020, 10, 307.
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212.
- Joerger, A.C.; Fersht, A.R. Structural Biology of the Tumor Suppressor p53. Annu. Rev. Biochem. 2008, 77, 557–582.
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355.
- Nicholls, C.D.; McLure, K.G.; Shields, M.A.; Lee, P.W.K. Biogenesis of p53 involves cotranslational dimerization of monomers and posttranslational dimerization of dimers. Implications on the dominant negative effect. J. Biol. Chem. 2002, 277, 12937–12945.
- Rajagopalan, S.; Huang, F.; Fersht, A.R. Single-molecule characterization of oligomerization kinetics and equilibria of the tumor suppressor p53. Nucleic Acids Res. 2011, 39, 2294–2303.
- Weinberg, R.L.; Veprintsev, D.B.; Fersht, A.R. Cooperative binding of tetrameric p53 to DNA. J. Mol. Biol. 2004, 341, 1145–1159.
- Sakaguchi, K.; Sakamoto, H.; Lewis, M.S.; Anderson, C.W.; Erickson, J.W.; Appella, E.; Xie, D. Phosphorylation of serine 392 stabilizes the tetramer formation of tumor suppressor protein p53. Biochemistry 1997, 36, 10117–10124.
- Rajagopalan, S.; Jaulent, A.M.; Wells, M.; Veprintsev, D.B.; Fersht, A.R. 14-3-3 activation of DNA binding of p53 by enhancing its association into tetramers. Nucleic Acids Res. 2008, 36, 5983–5991.
- Gaglia, G.; Guan, Y.; Shah, J.V.; Lahav, G. Activation and control of p53 tetramerization in individual living cells. Proc. Natl. Acad. Sci. USA 2013, 110, 15497–15501.
- Shakked, Z. Quaternary structure of p53: The light at the end of the tunnel. Proc. Natl. Acad. Sci. USA 2007, 104, 12231–12232.
- Wei, C.L.; Wu, Q.; Vega, V.B.; Chiu, K.P.; Ng, P.; Zhang, T.; Shahab, A.; Yong, H.C.; Fu, Y.T.; Weng, Z.; et al. A global map of p53 transcription-factor binding sites in the human genome. Cell 2006, 124, 207–219.
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412.
- McLure, K.G.; Lee, P.W.K. How p53 binds DNA as a tetramer. EMBO J. 1998, 17, 3342–3350.
- Kitayner, M.; Rozenberg, H.; Kessler, N.; Rabinovich, D.; Shaulov, L.; Haran, T.E.; Shakked, Z. Structural Basis of DNA Recognition by p53 Tetramers. Mol. Cell 2006, 22, 741–753.
- Tidow, H.; Melero, R.; Mylonas, E.; Freund, S.M.V.; Grossmann, J.G.; Carazo, J.M.; Svergun, D.I.; Valle, M.; Fersht, A.R. Quaternary structures of tumor suppressor p53 and a specific p53-DNA complex. Proc. Natl. Acad. Sci. USA 2007, 104, 12324–12329.
- Klein, C.; Planker, E.; Diercks, T.; Kessler, H.; Künkele, K.P.; Lang, K.; Hansen, S.; Schwaiger, M. NMR Spectroscopy Reveals the Solution Dimerization Interface of p53 Core Domains Bound to Their Consensus DNA. J. Biol. Chem. 2001, 276, 49020–49027.
- Wang, Y.; Schwedes, J.F.; Parks, D.; Mann, K.; Tegtmeyer, P. Interaction of p53 with its consensus DNA-binding site. Mol. Cell. Biol. 1995, 15, 2157–2165.
- Malecka, K.A.; Ho, W.C.; Ho, W.C. Crystal structure of a p53 core tetramer bound to DNA. Oncogene 2009, 28, 325–333.
- Stefan, M.I.; Le Novère, N. Cooperative Binding. PLoS Comput. Biol. 2013, 9, e1003106.
- Pauling, L. The Oxygen Equilibrium of Hemoglobin and Its Structural Interpretation. Proc. Natl. Acad. Sci. USA 1935, 21, 186–191.
- Jeffrey, P.D.; Gorina, S.; Pavletich, N.P. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science 1995, 267, 1498–1502.
- Balagurumoorthy, P.; Sakamoto, H.; Lewis, M.S.; Zambrano, N.; Clore, G.M.; Gronenborn, A.M.; Appella, E.; Harrington, R.E. Four p53 DNA-binding domain peptides bind natural p53-response elements and bend the DNA. Proc. Natl. Acad. Sci. USA 1995, 92, 8591–8595.
- Nagaich, A.K.; Zhurkin, V.B.; Sakamoto, H.; Gorin, A.A.; Clore, G.M.; Gronenborn, A.M.; Appella, E.; Harrington, R.E. Architectural accommodation in the complex of four p53 DNA binding domain peptides with the p21/waf1/cip1 DNA response element. J. Biol. Chem. 1997, 272, 14830–14841.
- Schlereth, K.; Beinoraviciute-Kellner, R.; Zeitlinger, M.K.; Bretz, A.C.; Sauer, M.; Charles, J.P.; Vogiatzi, F.; Leich, E.; Samans, B.; Eilers, M.; et al. DNA Binding Cooperativity of p53 Modulates the Decision between Cell-Cycle Arrest and Apoptosis. Mol. Cell 2010, 38, 356–368.
- Schlereth, K.; Heyl, C.; Krampitz, A.M.; Mernberger, M.; Finkernagel, F.; Scharfe, M.; Jarek, M.; Leich, E.; Rosenwald, A.; Stiewe, T. Characterization of the p53 Cistrome - DNA Binding Cooperativity Dissects p53′s Tumor Suppressor Functions. PLoS Genet. 2013, 9, e1003726.
- Rippin, T.M.; Freund, S.M.V.; Veprintsev, D.B.; Fersht, A.R. Recognition of DNA by p53 core domain and location of intermolecular contacts of cooperative binding. J. Mol. Biol. 2002, 319, 351–358.
- Dehner, A.; Klein, C.; Hansen, S.; Müller, L.; Buchner, J.; Schwaiger, M.; Kessler, H. Cooperative binding of p53 to DNA: Regulation by protein-protein interactions through a double salt bridge. Angew. Chem. Int. Ed. 2005, 44, 5247–5251.
- Veprintsev, D.B.; Freund, S.M.V.; Andreeva, A.; Rutledge, S.E.; Tidow, H.; Pérez Cañadillas, J.M.; Blair, C.M.; Fersht, A.R. Core domain interactions in full-length p53 in solution. Proc. Natl. Acad. Sci. USA 2006, 103, 2115–2119.
- Ho, W.C.; Fitzgerald, M.X.; Marmorstein, R. Structure of the p53 core domain dimer bound to DNA. J. Biol. Chem. 2006, 281, 20494–20502.
- Kitayner, M.; Rozenberg, H.; Rohs, R.; Suad, O.; Rabinovich, D.; Honig, B.; Shakked, Z. Diversity in DNA recognition by p53 revealed by crystal structures with Hoogsteen base pairs. Nat. Struct. Mol. Biol. 2010, 17, 423–429.
- Laptenko, O.; Shiff, I.; Freed-Pastor, W.; Zupnick, A.; Mattia, M.; Freulich, E.; Shamir, I.; Kadouri, N.; Kahan, T.; Manfredi, J.; et al. The p53 C Terminus Controls Site-Specific DNA Binding and Promotes Structural Changes within the Central DNA Binding Domain. Mol. Cell 2015, 57, 1034–1046.
- Enthart, A.; Klein, C.; Dehner, A.; Coles, M.; Gemmecker, G.; Kessler, H.; Hagn, F. Solution structure and binding specificity of the p63 DNA binding domain. Sci. Rep. 2016, 6.
- Ethayathulla, A.S.; Tse, P.W.; Monti, P.; Nguyen, S.; Inga, A.; Fronza, G.; Viadiu, H. Structure of p73 DNA-binding domain tetramer modulates p73 transactivation. Proc. Natl. Acad. Sci. USA 2012, 109, 6066–6071.
- Beno, I.; Rosenthal, K.; Levitine, M.; Shaulov, L.; Haran, T.E. Sequence-dependent cooperative binding of p53 to DNA targets and its relationship to the structural properties of the DNA targets. Nucleic Acids Res. 2011, 39, 1919–1932.
- Nagaich, A.K.; Zhurkin, V.B.; Durell, S.R.; Jernigan, R.L.; Appella, E.; Harrington, R.E. p53-Induced DNA bending and twisting: p53 Tetramer binds on the outer side of a DNA loop and increases DNA twisting. Proc. Natl. Acad. Sci. USA 1999, 96, 1875–1880.
- Sabapathy, K.; Lane, D.P. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 2017, 15, 13–30.
- Friedler, A.; Veprintsev, D.B.; Hansson, L.O.; Fersht, A.R. Kinetic instability of p53 core domain mutants. Implications for rescue by small molecules. J. Biol. Chem. 2003, 278, 24108–24112.
- Ang, H.C.; Joerger, A.C.; Mayer, S.; Fersht, A.R. Effects of common cancer mutations on stability and DNA binding of full-length p53 compared with isolated core domains. J. Biol. Chem. 2006, 281, 21934–21941.
- Bullock, A.N.; Henckel, J.; Fersht, A.R. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: Definition of mutant states for rescue in cancer therapy. Oncogene 2000, 19, 1245–1256.
- Joerger, A.C.; Ang, H.C.; Fersht, A.R. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl. Acad. Sci. USA 2006, 103, 15056–15061.
- Shiraishi, K.; Kato, S.; Han, S.Y.; Liu, W.; Otsuka, K.; Sakayori, M.; Ishida, T.; Takeda, M.; Kanamaru, R.; Ohuchi, N.; et al. Isolation of Temperature-sensitive p53 Mutations from a Comprehensive Missense Mutation Library. J. Biol. Chem. 2004, 279, 348–355.
- Dearth, L.R.; Qian, H.; Wang, T.; Baroni, T.E.; Zeng, J.; Chen, S.W.; Yi, S.Y.; Brachmann, R.K. Inactive full-length p53 mutants lacking dominant wild-type p53 inhibition highlight loss of heterozygosity as an important aspect of p53 status in human cancers. Carcinogenesis 2007, 28, 289–298.
- Paleček, E.; Ostatná, V.; Černocká, H.; Joerger, A.C.; Fersht, A.R. Electrocatalytic monitoring of metal binding and mutation-induced conformational changes in p53 at picomole level. J. Am. Chem. Soc. 2011, 133, 7190–7196.
- Ryan, K.M.; Vousden, K.H. Characterization of Structural p53 Mutants Which Show Selective Defects in Apoptosis but Not Cell Cycle Arrest. Mol. Cell. Biol. 1998, 18, 3692–3698.
- Ludwig, R.L.; Bates, S.; Vousden, K.H. Differential activation of target cellular promoters by p53 mutants with impaired apoptotic function. Mol. Cell. Biol. 1996, 16, 4952–4960.
- Timofeev, O.; Klimovich, B.; Schneikert, J.; Wanzel, M.; Pavlakis, E.; Noll, J.; Mutlu, S.; Elmshäuser, S.; Nist, A.; Mernberger, M.; et al. Residual apoptotic activity of a tumorigenic p53 mutant improves cancer therapy responses. EMBO J. 2019, 38, e102096.



