p53 can bind DNA in two modes: sequence-specifically via the DBD and in a sequence-independent manner through the CTD [
30]. Like many other transcription factors, p53 forms oligomers, and the transcriptionally active state of p53 requires the assembly of homotetramers [
31]. In vitro experiments suggested that p53 dimers are assembled co-translationally and are predominant under basal conditions [
32,
33,
34], whereas stress-induced post-translational modifications promote tetramerization [
35,
36]. A recent in vivo study has shown that in non-stressed cells monomers and dimers composed the major p53 pool, but DNA damage triggered rapid tetramerization [
37]. The ability of p53 to bind double-stranded DNA in a sequence-specific manner is determined by the DBD, which forms an immunoglobulin-like β-sandwich structure composed of a loop-sheet-helix motif and two large loops (L2 and L3) and serves as a basic scaffold for the DNA-binding surface. The loop-sheet-helix motif that includes the L1 loop binds the minor groove, whereas the L2 and L3 loops, stabilized by a zinc ion, dock to the major groove [
31,
38]. A typical p53 response element (RE) contains two decameric RRRCWWGYYY (R = A,G; W = A,T; Y = C,T) half-sites separated by spacers of 0–18 base pairs (bp) [
39,
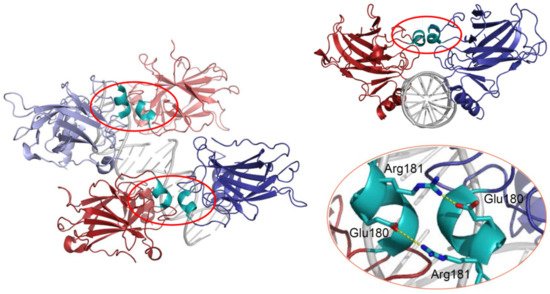
40]. Structural studies demonstrated that wild-type p53 proteins form tetrameric complexes on DNA, known as “dimers of dimers”, where each clamp-like symmetrical dimer binds one half-site of the RE, and two dimers occupy the full RE () [
31,
41,
42,
43,
44,
45,
46].
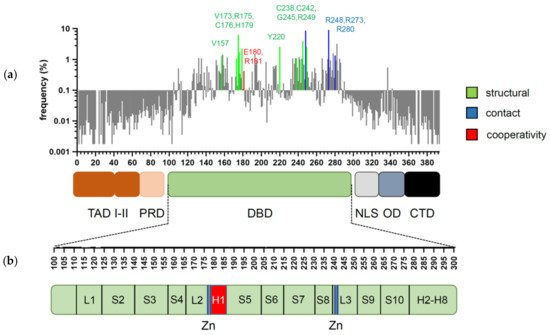
Assembly of p53 tetramers on DNA occurs in a highly cooperative manner. In general, cooperativity is a common biochemical phenomenon observed when a protein or protein complex contains multiple identical or near-identical ligand binding sites, and ligand binding to any one site increases (positive cooperativity) or decreases (negative cooperativity) the apparent affinity at the others. Cooperative binding can be mathematically described by the Hill equation, where the Hill coefficient functions as a quantitative measure of cooperativity [
47]. One of the best-known examples of positive cooperativity is oxygen binding by hemoglobin, where binding of each oxygen molecule increases the binding affinity for the next until the hemoglobin tetramer is fully saturated with oxygen at all four binding sites [
48]. Many other molecular assemblies exhibiting cooperative binding have been studied, including multimeric enzymes and transcription factors such as lambda phage repressor. In a very similar manner, DNA binding of p53 also displays positive cooperativity relying on the assembly of a tetrameric p53–DNA complex that is stabilized by protein–protein interactions between the four monomers [
41]. Primarily, tetramerization of p53 monomers is mediated by the OD, which consists of a short β-strand and an α-helix that provide an interaction surface for dimerization. Two primary dimers associate through their α-helices and build a four-helix bundle, stabilized by hydrophobic interactions [
49]. Integrity of the OD is essential for tetramerization and p53 functional activity. However, it has been shown that isolated DBDs upon interaction with p53 REs form tetramers also independent of the OD, although with 10- to 1000-fold lower binding affinity [
34,
50,
51]. These findings indicated existence of direct protein–protein interactions between DBDs. These interactions among p53 dimers and tetramers stabilize protein/DNA complexes and shape p53′s cistrome and transcriptional activity [
52,
53]. Early structural models of p53 DBD/DNA complexes pointed at the H1 helix as the structural basis for cooperative interactions between core domains [
31,
51]. Later models based on X-ray crystallography and NMR data indicated that the DBD interaction interface is formed by residues from the H1 helix (Pro177, His178, Glu180, Arg181) and several residues from the L3 loop (Met243, Gly244) [
44,
54]. The H1 helix is a short α-helical structure (Pro177-Cys182) located within the L2 loop, adjacent to the DNA-binding core region but not involved in direct contact with DNA (). Association of p53 with its RE brings the antiparallel oriented H1 helices of monomers in proximity and allows interactions that stabilize the entire complex. X-ray crystallography, NMR, and biochemical and biophysical studies pointed to an essential role of these structures for cooperative DNA binding and identified Glu180 and Arg181 as key residues that mediate reciprocal electrostatic interactions between H1 helices [
42,
44,
54,
55,
56,
57]. The double salt bridges formed by these oppositely charged amino acids maintain p53′s intra-dimer interactions and affect the strength of sequence-specific DNA binding [
52,
55,
56,
58]. Intriguingly, the CTD that binds DNA in a sequence-independent manner contributes to cooperativity by inducing conformational changes within the DBD that enhance sequence-specific binding [
59]. The primary structure of the H1 helix is highly conserved among p53 proteins of different vertebrate species and in the p53 family members p63 and p73, but the salt bridge itself is absent in p63 and p73 [
55,
57,
60,
61].
The degree of DNA binding cooperativity is determined by structural DNA properties encoded in the RE sequence—especially in its central WW dinucleotide [
62]. Even though these bases are not directly contacted by p53 residues, they are highly conserved and determine the torsional flexibility of the half-site and thus define the energy needed for DNA twisting [
62]. Since DNA binding of p53 induces significant RE bending and twisting, the exact WW dinucleotide sequence strongly affects binding [
50,
51,
57,
63]. In the case of a torsionally more flexible CATG, the p53 DBD binds with low cooperativity, while REs containing more rigid CAAG, and CTAG are bound with up to 3 orders of magnitude higher cooperativity and only when present in two contiguous p53 half-sites [
62]. Additionally, REs containing spacers between two half-sites are bound with high cooperativity [
62]. As transactivation relies on DNA binding and DNA-protein complex stability, cooperativity is a major determinant of p53′s transactivation function.