 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Isabel Desgagne-Penix | + 3884 word(s) | 3884 | 2021-05-17 08:30:00 | | | |
| 2 | Karina Chen | Meta information modification | 3884 | 2021-05-31 05:29:26 | | |
Video Upload Options
Alkaloids are an important group of specialized nitrogen metabolites with a wide range of biochemical and pharmacological effects. Since the first publication on lycorine in 1877, more than 650 alkaloids have been extracted from Amaryllidaceae bulbous plants and clustered together as the Amaryllidaceae alkaloids (AAs) family.
1. Classification of Amaryllidaceae Alkaloids
To date, more than 650 AAs have been reported, and their chemical library is still expanding [1][2][3][4][5][6][7][8][9][10][11][12][13][14]. Although diverse in structure, this plethora of AAs are categorized together as they share a common initial synthesis pathway. In previous literature, large numbers of AAs have been classified into different groups according to chemical characteristics, e.g., molecular skeleton and ring structure [1][15][16][17]. For this review, AAs were classified into 10 main groups instead, following a biochemical classification based on biogenetic lineage and ring type, to easily track the biosynthetic pathways [18] (Table 1, Figure 1). For example, haemanthamine and crinine were grouped together with respect to their biosynthetic origin and ring type even if they were previously categorized separately [19]. Some AAs with ring types different than those of group I to IX were classified in group X (or other-types) because they follow distinct biogenetic pathway, or because we cannot clearly indicate their biosynthetic origin (Table 1). Galanthindole contains a non-fused indole ring and might represent an artifact of homolycorine- or of pretazettine-type derivatives [20]. Ismine is considered to be a catabolic product from the haemanthamine-type skeleton, thus not a specific type of AA [21]. The latter examples demand further investigation on biogenetic origin and are not yet included on any particular type of AA.
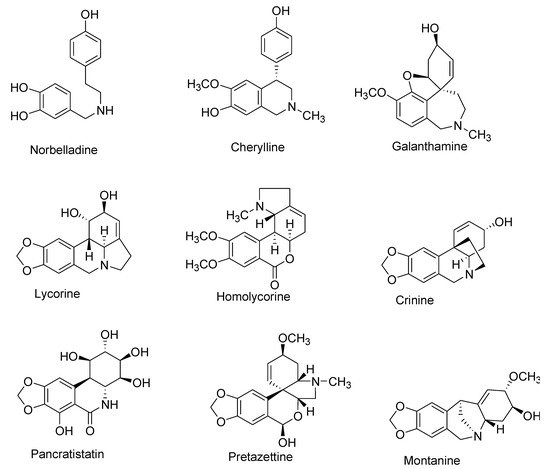
Figure 1. Representative Amaryllidaceae alkaloid structure for the main Amaryllidaceae alkaloid (AA)-types.
| Number | Type Name | Ring-Type |
|---|---|---|
| I | Norbelladine | N-(3,4-Dioxybenzyl)-4-oxyphenethylamine |
| II | Cherylline | Tetrahydroisoquinoline |
| III | Galantamine | 6H-Benzof,f]-2-benzazepine |
| IV | Lycorine | Pyrrolo[d,e]phenanthridine |
| V | Homolycorine | 2-Benzopyrano-[3,4-g]indole |
| VI | Crinine | 5,10b-Ethanophenanthridine |
| VII | Narciclasine | Lycoricidine |
| VIII | Pretazettine | 2-Benzopyrano [3,4-c]indole |
| IX | Montanine | 5,11-Methanomorphanthridine |
| X | Other | Different ring types and biogenetic origin |
Table 1. Main types of Amaryllidaceae alkaloids grouped according to their ring type and biosynthetic origin.
Some types of AA, such as plicamine and secoplicamine, are extracted in trace amounts exclusively from specific Amaryllidaceae species, such as Zephyranthes, but are classified in type X as they are rare, dinitrogenous members of AA, with a distinct biosynthetic linage [21][22][23][24]. Mesembrine alkaloids (also known as sceletium) have a distinct biosynthetic pathway, without norbelladine as key intermediate, they are usually extracted from Aizoaceae, but can be collected from several species of Amaryllidaceae [10]. Here, we concentrated exclusively on AAs that were discovered since 2015; hence, some alkaloids families are not represented.
2. Biosynthesis of Amaryllidaceae Alkaloids
Biosynthesis of AAs with their diverse and complex carbon skeleton involves a sequence of biochemical reactions such as oxidation, reduction, hydroxylation, methylation, phenol-phenol coupling, and oxide bridge formation. Although the complete AA biosynthetic pathway has not yet been elucidated, several steps with catalyzing enzymes can be predicted on the basis of the reaction types and enzyme families [25][18][26][27]. Here, we briefly discuss the AAs biosynthesis pathway and the enzymes involved.
Although novel AAs are still being discovered, radiolabeling experiments demonstrated that they all share a common biochemical pathway with a key intermediate; norbelladine, which is subsequently O-methylated, and then undergoes cyclization to give diverse basic skeletons of AAs (Figure 1; Figure 2) [28][29][30][31][32][33][34]. Norbelladine originates from the condensation of tyramine and 3,4-dihydroxybenzaldehyde (3,4-DHBA), molecules derived from the aromatic amino acids l-tyrosine and l-phenylalanine, respectively (Figure 2). The enzyme responsible for tyramine biosynthesis is the tyrosine decarboxylase (TYDC) (Figure 2). Two gene transcript variants of TYDC, named TYDC1 and TYDC2, were identified from the transcriptome of different Amaryllidaceae species including N. pseudonarcissus [35], Narcissus papyraceus [36], Lycoris radiata [37], and L. aureus [38].
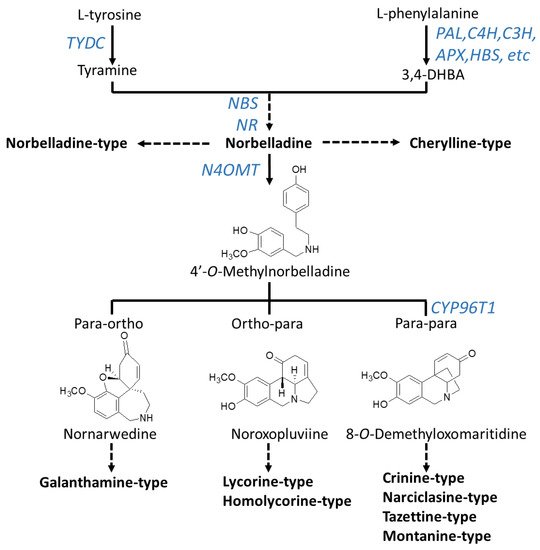
Figure 2. Biosynthesis pathway to major types of Amaryllidaceae alkaloids. Arrows without labeling reflect chemical reactions that have not been enzymatically characterized. Enzymes that have been identified are labeled in blue. A solid arrow symbolizes one enzymatic step whereas a broken arrow shows multiple enzymatic reactions. Chemical structures of precursors were added to clarify the regioselective phenol-phenol’ coupling reaction. Enzyme abbreviations: PAL, phenylalanine ammonia-lyase; C4H, cinnamate 4-hydroxylase; C3H, coumarate 3-hydroxylase; APX, ascorbate peroxidase; HBS, 4-hydroxybenzaldehyde synthase; TYDC, tyrosine decarboxylase; NBS, norbelladine synthase; NR, noroxomaritidine reductase; CYP96T1, cytochrome P450 monooxygenase 96T1.
The pathway leading to 3,4-DHBA from l-phenylalanine involves a series of reactions known as the phenylpropanoid pathway which is phylogenetically spread in most plant species. In Amaryllidaceae, using precursor feeding experiments, it was reported that trans-cinnamic acid, p-coumaric acid, and caffeic acid were intermediate products that ultimately led to 3,4-DHBA [39][40]. Specifically, l-phenylalanine is converted to trans-cinnamic acid by the phenylalanine ammonia-lyase (PAL) (Figure 2). Several PAL gene transcripts were identified and characterized from different species of Amaryllidaceae [35][36][37][41][42][43]. Interestingly, two main phylogenetic PAL clusters were identified; the first one contained PAL transcripts ubiquitously expressed in Amaryllidaceae whereas the second cluster contained PAL transcripts with expression highest and correlating with organs where AAs accumulated [18]. This indicates that different PAL transcripts encode enzymes with distinct functions in the phenylpropanoid pathway and it suggests the role of the latter cluster in AA biosynthesis. Next, trans-cinnamic acid is hydroxylated to form p-coumaric acid by the cinnamate 4-hydroxylase (C4H), a cytochrome P450 of the CYP73 subfamily (cinnamate 4-hydroxylase, C4H) [44][45]. Transcripts encoding C4H were reported from several Amaryllidaceae species [35][36][37][43] including C4H from L. radiate, which was characterized as producing a region-specific 4-hydroxylation of trans-cinnamic acid [43]. The reactions catalyzed by PAL and C4H are also crucial steps in the biosynthesis of many phenylpropanoids such as flavonoids, linins, coumarins and stilbenes. From there, the enzymes and order of reactions leading to 3,4-DHBA are not known however it is hypothesized that it may involve the CYP98A3 named coumarate 3-hydroxylase (C3H) and/or the ascorbate peroxidase (APX) and/or the 4-hydroxybenzaldehyde synthase (HBS) [18] (Figure 2). A few studies have reported on the presence of phenolic acids such as caffeic, p-coumaric, and ferulic acids in N. pseudonarcissus, N. poeticus and Galanthus nivalis [46][47][48]. In addition, 3,4-DHBA was detected in plants outside the Amaryllidaceae family [49]. Collectively, this suggest that the initial reactions and enzymes of the phenylpropanoid pathway participate in the synthesis of the AA precursor 3,4-DHBA.
The condensation of tyramine and 3,4-DHBA leads to the formation of norbelladine catalyzed by the norbelladine synthase (NBS) and/or the noroxomaritidine reductase (NR) or a combination of these enzymes [50][51]. Norbelladine is the precursor to all AAs. For example, norbelladine can undergo different biochemical reactions such as methylation, hydroxylation, dehydration, cyclization and tautomerization to form cherylline-type AAs (Figure 2). Alternatively, norbelladine can be methylated by the norbelladine 4′-O-methyltransferase (N4OMT) to form 4′-O-methylnorbelladine [52]. 4′-O-methylnorbelladine cyclization in a regioselective phenol-phenol oxidative coupling forms the ortho-para’, para-para’ or para-ortho’ C-C coupled producing the various structural types of AAs (Figure 2). The para-ortho’ C-C coupling leads to galantamine-type AAs whereas the ortho-para’ phenol coupling elaborates lycorine- and homolycorine-types of AAs (Figure 2). The para-para phenol-phenol’ coupling reaction produces the crinine-, narciclasine-, tazettine- and montanine-types of AAs. A cytochrome P450 sequence was identified through comparative transcriptomics of N. sp. aff. pseudonarcissus, Galanthus sp., and Galanthus elwesii. Through heterologous expression and characterization, CYP96T1 formed the products (10bR,4aS)-noroxomaritidine and (10bS,4aR)-noroxomaritidine from 4′-O-methylnorbelladine supporting its involvement as a para-para’ C-C phenol coupling cytochrome P450 [53].
The core skeletons obtained from norbelladine, methylnorbelladine, and the phenol coupling steps form the basis of AA diversity. A complex network of enzyme catalyzing various types of reactions, such as C-C and C-O bond formations, O- and N-methylations, demethylations, hydroxylations, oxidations and reductions, yield the several hundred of structurally related AAs. Only a few of these enzymes are known to date and they are reported in Figure 2.
3. Pharmacological Properties of Novel Amaryllidaceae Alkaloids
The pharmacological properties of the newly discovered AAs (1–91) were assessed when isolated in sufficient amount. AAs display a wide range of biological activities, including cytotoxicity, effects on the central nervous systems (CNS), anti-inflammatory, anti-microbial, anti-parasitic, larvicidal, and antioxidant activities.
3.1. Antitumoral Cytotoxic Activity
Lycorine is the most abundantly found AA, it belongs to the pyrrolo[de]phenanthridine subgroup. The biological effects of lycorine have been known for many years, and lycorine is still being investigated for a variety of therapeutic application, in particular as anticancer agent showing promising activity against tumors with dismal prognoses [54][55]. The structure–activity relationship (SAR) of lycorine and its derivatives has been evaluated using human leukemia T cells (Jurkat). The results showed that the free 1,2-diol functionality in the C-ring is required to induce apoptosis [56]. Furthermore, it has been demonstrated that the presence of the unaltered diol functionality in the C-ring in its original configuration in lycorine series, the stereochemistry of the C/D ring junction and the conformational freedom of the C-ring were required for the anticancer activity [54]. 4′-O,N-dimethylnorbelladine N-oxide (3) displayed a weak cytotoxicity activity against the human colon cancer cell line HCT116 at concentration ranging from 10−5 M and 10 −6 M [57].
Among the new lycorine-type alkaloids, (+)-1-hydroxy-ungeremine (17) was evaluated for its cytotoxic potential against BEN-MEN-1 (meningioma), CCF-STTG1 (astrocytoma), CHG-5 (glioma), SHG-44 (glioma), U251 (glioma), HL-60 (human myeloid leukemia), SMMC-7721 (hepatocellular carcinoma), and W480 (colon cancer) cell lines and exhibited the most potent cytotoxicity against all tested tumor cell lines except BEN-MEN-1, with IC50 values ranging from 9.4 to 11.8 μM [58]. Pseudolycorine N-oxide (25) was inactive against human cervical cancer (SiHa) and human epidermoid carcinoma (KB) cells [59].
Among the homolycorine-type alkaloids, (+)-2-hydroxy-8-demethyl-homolycorine-α-N-oxide (26) had no significant cytotoxic activity (IC50 > 80 μM) against BEN-MEN-1 (meningioma), CCF-STTG1 (astrocytoma), CHG-5 (glioma), SHG-44 (glioma), U251 (glioma), HL-60 (human myeloid leukemia), SMMC-7721 (hepatocellular carcinoma), and W480 (colon cancer) cell lines [58]. Lycoranines E and F (27 and 28) showed moderate cytotoxicity against A549 (human lung carcinoma) and LoVo (human colon carcinoma) cells lines with IC50 > 20 μM [60]. 2α-10bα-Dihydroxy-9-O-demethylhomolycorine (29) showed significant cytotoxicity against the HCT-116 (colon adenocarcinoma), OVCAR-8 (ovarian carcinoma) and SF-295 (glioblastoma) cell lines with IC50 values 11.69, 15.11, and 16.31 μM respectively [61].
Numerous additional types of AAs displayed interesting anti-cancer activity. Of the cherylline-type, gigantellinine (8) had a weak but significant cytotoxicity at 400 μM against breast cancer cell line MCF-7, while gigantelline (7) showed no cytotoxicity at the same concentration [62]. Crinine-derivatives (+)-6β-acetyl-8-hydroxy-9-methoxy-crinamine (31) showed significant cytotoxicity against HL-60 (IC50 < 10 μM), and moderate cytotoxicity against astrocytoma and glioma cell lines, CCF-STTG1, CHG-5, SHG- 44 and U251 (10 μM < IC50 ≤ 30 μM) [58]. The cleavage between C-1 and C-13 and the hydroxyl at C-6′ in crinine alkaloid skeleton might be essential to their bioactivity [63]. Crijaponine A (32) was inactive towards both human epithelial carcinoma HeLa and HL-60 cell lines (IC50 > 60 μM) [64]. 6α-Methoxyundulatine (33), 6α-methoxycrinamidine (34), and undulatine N-oxide (35) did not show significant cytotoxicity against the KB (derived from a human carcinoma of the naropharynx), HepG2 (human liver cancer), MCF7 (breast cancer), SK-Mel2 (melanoma), and LNCaP (human prostate) cancer cell lines with IC50 > 100 μM [65]. 1,4-Dihydroxy-3-methoxy powellan (36) displayed an IC50 > 60 μM against the A2780 human ovarian cancer cell line [66]. Gigancrinine (44) showed no cytotoxicity at 400 μM against breast cancer cell line MCF-7 [62]. 3-O-Methyl-epi-vittatine (49) did not shown any cytotoxicity against MCF7, TK10 and UACC62 cancer cell lines [67]. Narciclasine-4-O-β-d-xylopyranoside (51), a narciclasine-type was inactive against KB and SiHa cell lines at all the concentrations [68]. Jonquailine (52), a pretazettine-tye, showed significant antiproliferative activities against cells derived from glioblastoma (U87, U373, and Hs683), melanoma (SKMEL-28), uterine sarcoma (MES-SA and MES-SA/Dx5), and lung carcinoma (A549, H1993 and H2073) with IC50 values ranging from 1 to 85 μM [69]. Moreover, (52) showed synergic effects with paclitaxel in its anti-proliferative action against lung carcimoma drug-resistant H1993 and H2073 cells in a dose-dependent manner with IC50 values between 0.39 and 100 μM. The SAR of (52) suggested that hydroxylation of C-8 was required for its anticancer activity [69].
Among cripowellin-type AAs, 4,8-dimethoxy-cripowellin C (71), 4,8-dimethoxy-cripowellin D (72), 9-methoxy-cripowellin B (73), and 4-methoxy-8-hydroxy-cripowellin B (74) showed impressive cytotoxicity against seven lung cancer cell lines (A549, H446, H460, H292, 95-D, and SPCA-1) with IC50 < 30 nM, with (73) and (74) being more active than (71) and (72). Cripowellin C (75) and cripowellin D (76) were efficiently cytotoxic against the A2780 human ovarian cancer cell line with IC50 values of 25 ± 2, and 28 ± 1 nM, respectively [70].
Other-type of AA (+)-N-methoxylcarbonyl-2-demethyl-isocorydione (79) exhibited strong cytotoxic against all tested tumor cell lines (astrocytoma, glioma, human myeloid leukemia, hepatocellular carcinoma, colon cancer) except meningioma (BEN-MEN-1), with IC50 values of 9.2–12.8 μM [58]. Zephycandidine A (81) was cytotoxic for five cancer cell lines, HL-60, A549, MCF-7, colon cancer SW480 (colon cancer), and hepatocellular carcinoma SMMC- 7721 (hepatocellular carcinoma), with IC50 values of 1.98, 6.49, 3.44, 6.27, and 7.03 μM, respectively. Moreover, zephycandidine A (81) showed weak cytotoxicity against the normal Beas-2B cell line (IC50 = 20.08 μM) with selectivity index as high as 10 when compared normal Beas-2B cell line, via activation of caspase-3, upregulation of Bax, down-regulation of Bcl-2, and degradation of PARP expression [71]. Hymenolitatine (82) showed weak cytotoxic activity against four cancer cell lines, HepG-2, LoVo, Hela, and A549, with IC50 values of 75.19, 69.81, 96.37, and 102.53 μM, respectively [72]. Cripowellin-form (71–76) and Zephycandidine A (81) belonging to the other-type of AA may be potential targets for further anticancer investigations.
3.2. Effects on the Central Nervous System (CNS)
Several enzymes of the CNS are interesting drug targets. AChE is a serine protease located at neuromuscular junctions, in cholinergic synapses of the central nervous system and in red blood cells [73][74][75]. The enzyme catalyzes the rapid hydrolysis and inactivation of the neurotransmitter acetylcholine into acetate and choline to enable cholinergic neurons to return to their resting state. Butyrylcholinesterase (BChE) can also hydrolyze acetylcholinesterase into acetate and choline. BChE is produced by the liver and detected in the plasma. Changes in its plasmatic levels can indicate of liver dysfunction. BChE is also expressed in neurons of the CNS [76].
In Alzheimer’s disease (AD), AChE is overly active, and the consequential lower level of acetylcholine in the brain cause weakened neurotransmission [77]. Similarly, BChE deregulation is measured in the brain of individuals suffering from AD. Malfunction of the cholinergic system may be pharmacologically tackled via AChE inhibitors that ameliorate the cholinergic deficit at early stages of the disease and reduce progression. In addition, glycogen synthase kinase-3 (GSK-3) is a ubiquitous serine/threonine kinase, implicated in AD, which can trigger abnormal hyperphosphorylation of tau protein, which is believed to be a critical event in neurofibrilary tangle formation. Thus, GSK-3 inhibition represents an attractive drug target for AD and other neurodegenerative disorders [78]. Finally, prolyloligopeptidase (POP) is a cytosolic serine peptidase widely distributed in the organs of the body, including the brain, which cleaves peptide bonds at the carboxyl end of proline [79][80]. Previous studies have shown that POP inhibitors are efficient anti-dementia drugs [81][82].
The AA galantamine, donepezil and rivastigmine are potent reversible inhibitors of AChE approved for the symptomatic treatment of AD [83][84]. Since cholinesterase enzyme inhibitors are first generation drugs for AD, AChE and BChE are the most targeted enzymes at the moment.
Galantamine derivative sanguinine is ten times more active than galantamine whereas 11-hydroxygalantamine exhibits inhibitory activity similar to that of galantamine. The extra or protected hydroxyl group in its allylic position in (R1) may be required for the activities [85]. SAR of galantamine and its derivatives was comprehensively reviewed elsewhere [16].
Among the six new galantamine-type alkaloids only 9-de-O-methyl-11β-hydroxygalantamine (13) showed a weak AChE inhibitory activity with IC50 value 168.7 μM. The SAR of new galantamine derivatives alkaloids (11–16) and known alkaloids isolated from the same plant species revealed that the 4,4a double bond and 9-OH are required for the AChE inhibitory activity, while the presence of the 11-OH group dramatically decreases AChE inhibitory activity [86].
Norbelladine-type alkaloids 6-O-demethylbelladine (1) and 4′-O-demethylbelladine (2) were identified as weak inhibitors of AChE with IC50 values of 223.2 ± 23.6 and 606.8 ± 74.2 μM, respectively, for (1) and (2) [87]. They were more potent against BChE, (2) (IC50 = 30.7 ± 4.0 μM) being more active than (1) (IC50 = 115.7 ± 10.1μM). 4′-O-Demethylbelladine (2) exhibited weak POP inhibition with IC50 value of 370 ± 30 μM), but more potently than 6-O-demethylbelladine (1) (IC50 = 660 ± 90 μM) [87]. Carltonine A–C (4–6) were evaluated for their potential activity against AChE, BChE and POP. Carltonine A–B (4 and 5) inhibited BChE with IC50 values of 0.91 ± 0.02 and 0.031 ± 0.001 μM. Computational studies detected a plausible binding site on BChE while SAR suggested that the 1,3-dioxolane ring of carltonine B of (5) may be responsible of the BChE activity compared to the opening dimethoxybenzene analogue of (4) [88].
As for the cherylline-type, gigantellinine (8) inhibited the activity of AChE in a dose-dependent manner with IC50 value of 174.90 ± 2.30 μM, while gigantelline (7) did not [62].
The lycorine-type, galanthine N-β-oxide (20) did not exhibit significantly AChE inhibitory activity (IC50 > 200 μM) compared to galanthine. The N-oxide fragment seems to be deactivating the inhibitory activity of AChE. New lycorine derivatives (22) displayed moderate AChE inhibition (IC50 = 35.61 ± 1.90 μM) compared to carinatine (IC50 > 200 μM). The aromatic C-ring in lycorine-sort alkaloids may be essential for their activity against AChE [89]. Oxoincartine (23) was inactive against both cholinesterases (AChE and BChE) [90].
7-Hydroxyclivonine (30), a homolycorine-type exhibited weak AChE inhibition with IC50 value 114.07 μM and moderate effect against BChE with IC50 = 67.3 μM. Molecular docking with BChE revealed that (30) and galantamine both interact with similar amino acids in the same binding pocket [91].
In the crinine subgroup, augustine N-oxide (37) showed moderate inhibition of AChE at 79.64 μg·mL−1 (250.44 μM), while buphanisine N-oxide (38) did not inhibit the enzyme activity [92]. A mixture of 6α-hydroxymaritidine (39) and 6β-hydroxymaritidine (40) showed weak AChE inhibitory activity with IC50 = 90.43 μM, and was inactive of for BChE inhibitory activity (IC50 > 600 μM). Molecular docking studies of 6α-hydroxymaritidine (39), 6β-hydroxymaritidine (40), lycorine-type reticulinine (18), and isoreticulinine (19) at the active sites of AChE and BChE identified (19) as a potential inhibitory molecule, since it was stabilized in the active site through hydrogen bonds, π-π stacking and hydrophobic interactions [93]. Crijaponine A (32) showed anti-AChE activity with IC50 > 10 μM [64]. Moreover, 11-O-Acetyl-9-O-demethylmaritidine (42) inhibited both cholinesterases (AChE and BChE) with IC50 values 6.04 and 29.72 μM, respectively. Furthermore, 3,11-O-Diacetyl-9-O-demethylmaritidine (41) showed weaker AChE inhibition with IC50 value 67.4 μM and was inactive against BChE. The SAR of some haemanthamine-derivatives revealed that two free hydroxyl groups present at C-3 and C-11 may be essential for the anti-cholinesterase activity [90]. Gigancrinine (44) did not show any anti-AChE activity [62].
The 4-O-Methylnangustine (55), a montanine-type AA was inactive (IC50 > 200 μM) against both AChE and BChE [91].
Among plicamine- and seco-plicamine subgroups of alkaloids, (61–63) exhibited weak AChE inhibition, with IC50 values of 110.6, 57.26, and 75.3 μM [86]. Moreover, (67) showed weak AChE inhibitory activity with IC50 value 126.16 μM, whereas the others new plicamine- and seco-plicamine-types were almost inactive (IC50 > 200 μM). The N-2-hydroxyethyl group in plicamine-form alkaloids may be essential for this activity [89].
Other types of alkaloids, zephycandidine III (85) significantly inhibited AChE with IC50 value of 8.82 μM, while (83) and (84) were inactive at 200 μM. Narcipavline (86) exhibited weak AChE inhibitory activity with IC50 value 208 ± 37 μM and a significant BChE inhibitory activity IC50 = 24.4 ± 1.2 μM. Zephycandidine A (81) displayed anti-AChE activity in a dose-dependent manner with IC50 = 127.99 μM. Molecular docking studies of zephycandidines (81), (83–85) and galantamine with AChE revealed that interactions with W286 and Y337 are necessary for inhibitory activity [23] [71]. Narcimatuline (88) was evaluated for its AChE, BChE, POP, and GSK-3β inhibitory activities and significant inhibited BChE, POP and GSK-3 activities, with IC50 values of 5.9 ± 0.2, 29.2 ± 1.0 and 20.7 ± 2.4 μM, respectively, but only a weak activity against AChE with IC50 value 489 ± 60 μM [94].
Of all the candidates investigated recently, crinine-derivative 11-O-acetyl-9-O-demethylmaritidine (42) and other-type Zephycandidine III (85) are interesting targets for further anti-AChE investigation for Alzheimer’s disease.
3.3. Anti-Inflammatory and Antioxidant Activity
Unfortunately, the anti-inflammatory activity of AAs is rarely reported. In vitro anti-inflammatory studies of fifteen AAs isolated from different Crinum species was comprehensively reviewed in 2003 and their activity was very low [95]. More recently, the anti-inflammatory activity of lycorine-type AAs, such as (+)-1-hydroxy-ungeremine (17), homolycorine-type, such as (+)-2-hydroxy-8-demethyl-homolycorine-α-N-oxide (26), crinine-type (+)-6β-acetyl-8-hydroxy-9-methoxy-crinamine (31), and other-type (+)-N-methoxylcarbonyl-2-demethyl-isocorydione (79) against cyclooxygenase-1 (COX,-1), and cyclooxygenase-2 (COX-2) was evaluated in vitro, and AA (17) and (79) displayed selective inhibition of COX-2 (>90%) [58].
Cripowellin derivatives, 4,8-dimethoxy-cripowellin C (71), 4,8-dimethoxy-cripowellin D (72), 9-methoxy-cripowellin B (73) and 4-methoxy-8-hydroxy-cripowellin B (74) displayed significant inhibition of COX-1 (>64%) and of COX-2 (>90%), respectively [63]. They were also evaluated for their antioxidant potential activities using ABTS·+ (2,2′-azino-bis(3-ethylbenzo-thiazoline-6-sulphonic acid) and DPPH (1,1-diphenyl-2-picrylhydrazyl) methods. AAs (72), (73), and (74) showed significant antiradical activity with IC50 values of from 52.2 to 80.1μM.
The anti-inflammatory activity of galantamine-, plicamin- and seco-plicamin-type (11−16, 56–67 and 68–69) was evaluated in vitro by studying the inhibition of lipopolysaccharide (LPS)-induced nitric oxide (NO) production in RAW 264.7 mouse macrophages. Among the tested compounds, two plicamine-type alkaloids (59) and (60) showed significant inhibitory activities with IC50 values of 18.77 and 10.21 μM, respectively, while other alkaloids were inactive at 200 μM [86].
3.4. Anti-Parasitic and Antibacterial Activity
The 4,8-Dimethoxy-cripowellin C (71), 4,8-dimethoxy-cripowellin D (72), 9-methoxy-cripowellin B (73), and 4-methoxy-8-hydroxy-cripowellin B (74) were evaluated for their antimicrobial against eight species of bacteria (Streptococcus pneumoniae, Staphylococcus aureus, Staphylococcus epidermidis, Klebsiella pneumoniae, Pseudomonas aeruginosa, Haemophilus influenzae, Enterobacter cloacae, and Shigella dysenteriae). Moreover, (73) and (74) displayed highest antibacterial activity with IC50 values < 0.50 mM, while (71) and (72) had weak activity [63].
Malaria (Plasmodium sp.), leishmaniasis (Leishmania sp.), and trypanosomiasis (Trypanosoma brucei and Trypanosoma cruzi) are the most common chronic protozoan diseases and occur mainly in poor rural and urban areas in tropical and subtropical regions of the world. Previously, several AAs were reported for their potent in vitro antiprotozoal activity [96]. The anti-plasmodial activity was recently reviewed elsewhere [97][98]. Newly isolated alkaloids such as cripowellin C (75) and D (76) were evaluated against the chloroquine/mefloquine-resistant Dd2 strain of Plasmodium falciparum and were found to have potent antiplasmodial activity, with IC50 values of 180 ± 20, 26 ± 2, and 260 ± 20 nM, respectively [70].
Crinine-type 1,4-dihydroxy-3-methoxy powellan (36) had a weak activity, with IC50 = 37 ± 3 μM against the same pathogen [66]. The two epimers (6α-hydroxymaritidine (39) and 6β-hydroxymaritidine (40)) were evaluated for their antiprotozoal activity against T. brucei rhodesiense (trypomastigotes forms, STIB 900 strain), T. cruzi (amastigotes forms, Tulahuen C4 strain), L. donovani (amastigotes forms, MHOM-ET-67/L82 strain), and P. falciparum (intraerythrocytic forms, IEF, NF54 strain). They displayed low toxicity against all protozoans tested with IC50 values of 30.68, 66.11, > 100 and 32.86 μg.mL−1 respectively [93]. Augustine N-oxide (37) and buphanisine N-oxide (38) were also evaluated against the same strains mentioned above and displayed low activity with IC50 values ranging from 32 to > 100 μg.mL−1. The presence of an N-oxide group in (37) and (38) appears to decrease their activity against T. brucei and P. falciparum compared to the previously characterized anti-protozoal compounds belonging to the same subgroup [92].
Scillitazettine (53) and scilli-N-desmethylpretazettine (54) were evaluated against the chloroquine-resistant strain P. falciparum FcB1 and displayed antiparasitic activity with IC50 values of 77.0 ± 2.0 and 46.5 ± 2.0 μM, respectively [57].
3.5. Larvicidal and Insecticidal
Insects are important vectors of many diseases, controlling their proliferation is an efficient way of preventing disease spread. Aedes aegypti is the main vector for dengue, yellow fever and Zika infection. In an earlier study, organic extracts of the bulbs of Nerine sarniensis, demonstrated strong larvicidal and insecticidal activity with LC50 of 0.008 μg.μL−1 against A. aegypti larvae and against grown-up females with LD50 of 4.6 μg/mosquito. Sarniensine (78) was less efficient against larvae at the most minimal concentration of 0.1 μg/μL but displayed strong adulticidal activity with an LD50 of 1.38 ± 0.056 μg/mosquito [99]. Mesembrine-class sarniensinol (77), sarniensine (78) and crinine-type crinsarnine (43) had no effect against A. aegypti larvae at all concentrations tested. In adult topical bioassays, only (43) displayed adulticidal activity, with an LD50 = 2.29 ± 0.049 μg per mosquito. SAR studies revealed that the scaffold of pretazettine alkaloids in (77) and (78) and (43) and in bowdensine were important for their activity. Among the mesembrine group, the opening of the B-ring or the presence of a B-ring lactone as well as the trans-stereochemistry of the A/B-ring junction seem to be important for their activity, while in crinine-type alkaloids, the substituent at C-2 appears to be important [100][99][101].
3.6. Others Activities
The carbonic anhydrases (CAs) are metalloenzymes that catalyze the reversible hydration of carbon dioxide with water into a bicarbonate ion and a proton. In humans, sixteen isozymes have been identified including human cabonic isozyme II (hCAII) reported to be involved many diseases like glaucoma, epilepsy and cancer. Thus, hCAII is a target for therapeutic interventions [102]. Crinasiaticine A (46) and crinasiaticine B (47) were evaluated for their inhibitory potential against hCAII and they were inactive [102].
References
- Jin, Z.; Yao, G. Amaryllidaceae and Sceletium alkaloids. Nat. Prod. Rep. 2019, 36, 1462–1488.
- Lewis, J.R. Amaryllidaceae and Sceletium alkaloids. Nat. Prod. Rep. 1992, 9, 183–191.
- Lewis, J.R. Amaryllidaceae and Sceletium alkaloids. Nat. Prod. Rep. 1993, 10, 291–299.
- Lewis, J.R. Amaryllidaceae and Sceletium alkaloids. Nat. Prod. Rep. 1995, 12, 339–345.
- Lewis, J.R. Amaryllidaceae and Sceletium alkaloids. Nat. Prod. Rep. 1996, 13, 171–176.
- Lewis, J.R. Amaryllidaceae, sceletium, imidazole, oxazole, thiazole, peptide and miscellaneous alkaloids. Nat. Prod. Rep. 2002, 19, 223–258.
- Jin, Z.; Li, Z.; Huang, R. Muscarine, imidazole, oxazole, thiazole, Amaryllidaceae and Sceletium alkaloids. Nat. Prod. Rep. 2002, 19, 454–476.
- Jin, Z. Amaryllidaceae and Sceletium alkaloids. Nat. Prod. Rep. 2003, 20, 606–614.
- Jin, Z. Amaryllidaceae and Sceletium alkaloids. Nat. Prod. Rep. 2005, 22, 111–126.
- Jin, Z. Amaryllidaceae and Sceletium alkaloids. Nat. Prod. Rep. 2007, 24, 886–905.
- Jin, Z. Amaryllidaceae and Sceletium alkaloids. Nat. Prod. Rep. 2009, 26, 363–381.
- Jin, Z. Amaryllidaceae and Sceletium alkaloids. Nat. Prod. Rep. 2011, 28, 1126–1142.
- Jin, Z. Amaryllidaceae and Sceletium alkaloids. Nat. Prod. Rep. 2013, 30, 849–868.
- Jin, Z. Amaryllidaceae and Sceletium alkaloids. Nat. Prod. Rep. 2016, 33, 1318–1343.
- Kornienko, A.; Evidente, A. Chemistry, biology, and medicinal potential of narciclasine and its congeners. Chem. Rev. 2008, 108, 1982–2014.
- He, M.M.; Qu, C.R.; Gao, O.D.; Hu, X.M.; Hong, X.C. Biological and pharmacological activities of Amaryllidaceae alkaloids. Rsc. Adv. 2015, 5, 16562–16574.
- Ding, Y.; Qu, D.; Zhang, K.M.; Cang, X.X.; Kou, Z.N.; Xiao, W.; Zhu, J.B. Phytochemical and biological investigations of Amaryllidaceae alkaloids: A review. J. Asian Nat. Prod. Res. 2017, 19, 53–100.
- Desgagné-Penix, I. Biosynthesis of alkaloids in Amaryllidaceae plants: A review. Phytochem. Rev. 2020.
- Berkov, S.; Osorio, E.; Viladomat, F.; Bastida, J. Chemodiversity, chemotaxonomy and chemoecology of Amaryllidaceae alkaloids. Alkaloids Chem. Biol. 2020, 83, 113–185.
- Unver, N.; Kaya, G.I.; Werner, C.; Verpoorte, R. Galanthindole: A new indole alkaloid from Galanthus plicatus ssp. byzantinus. Planta. Med. 2003, 69, 869–871.
- Bastida, J.; Berkov, S.; Torras, L.; Pigni, N.B.; de Andrade, J.P.; Martinez, V.; Codina, C.; Viladomat, F. Chemical and biological aspects of Amaryllidaceae alkaloids. Rec. Adv. Pharm. Sci. 2011, 65–100.
- Safratova, M.; Hostalkova, A.; Hulcova, D.; Breiterova, K.; Hrabcova, V.; Machado, M.; Fontinha, D.; Prudencio, M.; Kunes, J.; Chlebek, J.; et al. Alkaloids from Narcissus poeticus cv. Pink Parasol of various structural types and their biological activity. Arch. Pharm. Res. 2018, 41, 208–218.
- Zhan, G.; Liu, J.; Zhou, J.; Sun, B.; Aisa, H.A.; Yao, G. Amaryllidaceae alkaloids with new framework types from Zephyranthes candida as potent acetylcholinesterase inhibitors. Eur. J. Med. Chem. 2017, 127, 771–780.
- Wang, H.Y.; Qu, S.M.; Wang, Y.; Wang, H.T. Cytotoxic and anti-inflammatory active plicamine alkaloids from Zephyranthes grandiflora. Fitoterapia 2018, 130, 163–168.
- Singh, A.; Desgagné-Penix, I. Biosynthesis of the Amaryllidaceae alkaloids. Plant. Sci. Today 2014, 1, 114–120.
- Singh, A.; Desgagné-Penix, I. Chapter 3: Biosynthesis of Amaryllidaceae Alkaloids: A Biochemical Outlook. In Alkaloids: Biosynthesis, Biological Roles and Health Benefits; Sobarzo-Sanchez, E., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2015.
- Kilgore, M.B.; Kutchan, T.M. The Amaryllidaceae alkaloids: Biosynthesis and methods for enzyme discovery. Phytochem. Rev. 2016, 15, 317–337.
- El Tahchy, A.; Ptak, A.; Boisbrun, M.; Barre, E.; Guillou, C.; Dupire, F.; Chretien, F.; Henry, M.; Chapleur, Y.; Laurain-Mattar, D. Kinetic study of the rearrangement of deuterium-labeled 4′-O-methylnorbelladine in Leucojum aestivum shoot cultures by mass spectrometry. Influence of precursor feeding on amaryllidaceae alkaloid accumulation. J. Nat. Prod. 2011, 74, 2356–2361.
- Saliba, S.; Ptak, A.; Laurain-Mattar, D. 4′-O-Methylnorbelladine feeding enhances galantamine and lycorine production by Leucojum aestivum L. shoot cultures. Eng. Life Sci. 2015, 15, 640–645.
- Barton, D.H.R.; Kirby, G.W. Phenol oxidation and biosynthesis. Part V. The synthesis of galantamine. J. Chem. Soc. (Resumed) 1962, 153, 806–817.
- Barton, D.H.R.; Kirby, G.W.; Taylor, J.B.; Thomas, G.M. Phenol oxidation and biosynthesis. Part VI. The biogenesis of Amaryllidaceae alkaloids. J. Chem. Soc. (Resumed) 1963, 866, 4545–4558.
- Eichhorn, J.; Takada, T.; Kita, Y.; Zenk, M.H. Biosynthesis of the Amaryllidaceae alkaloid galantamine. Phytochemistry 1998, 49, 1037–1047.
- El Tahchy, A. Étude de la voie de biosynthèse de la galantamine chez Leucojum aestivum L.—Criblage phytochimique de quelques Amaryllidaceae. Ph.D. Thesis, Nancy Université Henri Poincaré, Nancy, France, 2010.
- El Tahchy, A.; Boisbrun, M.; Ptak, A.; Dupire, F.; Chretien, F.; Henry, M.; Chapleur, Y.; Laurain-Mattar, D. New method for the study of Amaryllidaceae alkaloid biosynthesis using biotransformation of deuterium-labeled precursor in tissue cultures. Acta. Biochim. Pol. 2010, 57, 75–82.
- Singh, A.; Desgagne-Penix, I. Transcriptome and metabolome profiling of Narcissus pseudonarcissus ‘King Alfred’ reveal components of Amaryllidaceae alkaloid metabolism. Sci. Rep. 2017, 7, 17356.
- Hotchandani, T.; de Villers, J.; Desgagne-Penix, I. Developmental Regulation of the Expression of Amaryllidaceae Alkaloid Biosynthetic Genes in Narcissus papyraceus. Genes 2019, 10, 594.
- Park, C.H.; Yeo, H.J.; Park, Y.E.; Baek, S.A.; Kim, J.K.; Park, S.U. Transcriptome Analysis and Metabolic Profiling of Lycoris radiata. Biology 2019, 8, 63.
- Wang, R.; Han, X.; Xu, S.; Xia, B.; Jiang, Y.; Xue, Y.; Wang, R. Cloning and characterization of a tyrosine decarboxylase involved in the biosynthesis of galantamine in Lycoris aurea. PeerJ 2019, 7, e6729.
- Suhadolnik, R.J.; Fischer, A.G.; Zulalian, J. Biogenesis of the Amaryllidaceae alkaloids. II. Studies with whole plants, floral primordia and cell free extracts. Biochem. Biophys. Res. Commun. 1963, 11, 208–212.
- Wildman, W.; Battersby, A.; Breuer, S. Biosynthesis in the Amaryllidaceae. Incorporation of 3-C14-Tyrosine and Phenylalanine in Nerine Bowdenii W. Wats. J. Am. Chem. Soc. 1962, 84, 4599–4600.
- Jiang, Y.; Xia, N.; Li, X.; Shen, W.; Liang, L.; Wang, C.; Wang, R.; Peng, F.; Xia, B. Molecular cloning and characterization of a phenylalanine ammonia-lyase gene (LrPAL) from Lycoris radiata. Mol. Biol. Rep. 2011, 38, 1935–1940.
- Jiang, Y.; Xia, B.; Liang, L.; Li, X.; Xu, S.; Peng, F.; Wang, R. Molecular and analysis of a phenylalanine ammonia-lyase gene (LrPAL2) from Lycoris radiata. Mol. Biol. Rep. 2013, 40, 2293–2300.
- Li, W.; Yang, Y.; Qiao, C.; Zhang, G.; Luo, Y. Functional characterization of phenylalanine ammonia-lyase- and cinnamate 4-hydroxylase-encoding genes from Lycoris radiata, a galantamine-producing plant. Int. J. Biol. Macromol. 2018, 117, 1264–1279.
- Fahrendorf, T.; Dixon, R.A. Stress responses in alfalfa (Medicago sativa L.). XVIII: Molecular cloning and expression of the elicitor-inducible cinnamic acid 4-hydroxylase cytochrome P450. Arch. Biochem. Biophys. 1993, 305, 509–515.
- Teutsch, H.G.; Hasenfratz, M.P.; Lesot, A.; Stoltz, C.; Garnier, J.M.; Jeltsch, J.M.; Durst, F.; Werck-Reichhart, D. Isolation and sequence of a cDNA encoding the Jerusalem artichoke cinnamate 4-hydroxylase, a major plant cytochrome P450 involved in the general phenylpropanoid pathway. Proc. Natl. Acad. Sci. USA 1993, 90, 4102–4106.
- Nikolova, M.; Gevrenova, R. Determination of phenolic acids in amaryllidaceae species by high performance liquid chromatography. Pharm. Biol. 2005, 43, 289–291.
- Benedec, D.; Oniga, I.; Hanganu, D.; Gheldiu, A.M.; Puscas, C.; Silaghi-Dumitrescu, R.; Duma, M.; Tiperciuc, B.; Varban, R.; Vlase, L. Sources for developing new medicinal products: Biochemical investigations on alcoholic extracts obtained from aerial parts of some Romanian Amaryllidaceae species. BMC Complement. Altern Med. 2018, 18, 226.
- Ferdausi, A.; Chang, X.M.; Hall, A.; Jones, M. Galantamine production in tissue culture and metabolomic study on Amaryllidaceae alkaloids in Narcissus pseudonarcissus cv. Carlton. Ind. Crops Prod. 2020, 144, 112058.
- Prachayasittikul, S.; Buraparuangsang, P.; Worachartcheewan, A.; Isarankura-Na-Ayudhya, C.; Ruchirawat, S.; Prachayasittikul, V. Antimicrobial and antioxidative activities of bioactive constituents from Hydnophytum formicarum Jack. Molecules 2008, 13, 904–921.
- Singh, A.; Massicotte, M.A.; Garand, A.; Tousignant, L.; Ouellette, V.; Berube, G.; Desgagne-Penix, I. Cloning and characterization of norbelladine synthase catalyzing the first committed reaction in Amaryllidaceae alkaloid biosynthesis. BMC Plant. Biol. 2018, 18, 338.
- Kilgore, M.B.; Holland, C.K.; Jez, J.M.; Kutchan, T.M. Identification of a Noroxomaritidine Reductase with Amaryllidaceae Alkaloid Biosynthesis Related Activities. J. Biol. Chem. 2016, 291, 16740–16752.
- Kilgore, M.B.; Augustin, M.M.; Starks, C.M.; O’Neil-Johnson, M.; May, G.D.; Crow, J.A.; Kutchan, T.M. Cloning and characterization of a norbelladine 4′-O-methyltransferase involved in the biosynthesis of the Alzheimer’s drug galantamine in Narcissus sp. aff. pseudonarcissus. PLoS ONE 2014, 9, e103223.
- Kilgore, M.B.; Augustin, M.M.; May, G.D.; Crow, J.A.; Kutchan, T.M. CYP96T1 of Narcissus sp. aff. pseudonarcissus Catalyzes Formation of the Para-Para’ C-C Phenol Couple in the Amaryllidaceae Alkaloids. Front. Plant. Sci. 2016, 7, 225.
- Lamoral-Theys, D.; Andolfi, A.; Van Goietsenoven, G.; Cimmino, A.; Le Calve, B.; Wauthoz, N.; Megalizzi, V.; Gras, T.; Bruyere, C.; Dubois, J.; et al. Lycorine, the main phenanthridine Amaryllidaceae alkaloid, exhibits significant antitumor activity in cancer cells that display resistance to proapoptotic stimuli: An investigation of structure-activity relationship and mechanistic insight. J. Med. Chem. 2009, 52, 6244–6256.
- Lamoral-Theys, D.; Decaestecker, C.; Mathieu, V.; Dubois, J.; Kornienko, A.; Kiss, R.; Evidente, A.; Pottier, L. Lycorine and its derivatives for anticancer drug design. Mini. Rev. Med. Chem. 2010, 10, 41–50.
- McNulty, J.; Nair, J.J.; Bastida, J.; Pandey, S.; Griffin, C. Structure-activity studies on the lycorine pharmacophore: A potent inducer of apoptosis in human leukemia cells. Phytochemistry 2009, 70, 913–919.
- N’Tamon, A.D.; Okpekon, A.T.; Bony, N.F.; Bernadat, G.; Gallard, J.-F.; Kouamé, T.; Séon-Méniel, B.; Leblanc, K.; Rharrabti, S.; Mouray, E. Streamlined targeting of Amaryllidaceae alkaloids from the bulbs of Crinum scillifolium using spectrometric and taxonomically-informed scoring metabolite annotations. Phytochemistry 2020, 179, 112485.
- Liu, Z.M.; Huang, X.Y.; Cui, M.R.; Zhang, X.D.; Chen, Z.; Yang, B.S.; Zhao, X.K. Amaryllidaceae alkaloids from the bulbs of Lycoris radiata with cytotoxic and anti-inflammatory activities. Fitoterapia 2015, 101, 188–193.
- Katoch, D.; Kumar, D.; Padwad, Y.S.; Singh, B.; Sharma, U. Pseudolycorine N-oxide, a new N-oxide from Narcissus tazetta. Nat. Prod. Res. 2020, 34, 2051–2058.
- Ang, S.; Liu, X.M.; Huang, X.J.; Zhang, D.M.; Zhang, W.; Wang, L.; Ye, W.C. Four New Amaryllidaceae Alkaloids from Lycoris radiata and Their Cytotoxicity. Planta Med. 2015, 81, 1712–1718.
- Carvalho, K.R.; Silva, A.B.; Torres, M.C.M.; Pinto, F.C.L.; Guimaraes, L.A.; Rocha, D.D.; Silveira, E.R.; Costa-Lotufo, L.V.; Braz, R.; Pessoa, O.D.L. Cytotoxic Alkaloids from Hippeastrum solandriflorum Lindl. J. Braz. Chem. Soc. 2015, 26, 1976–1980.
- Ka, S.; Masi, M.; Merindol, N.; Di Lecce, R.; Plourde, M.B.; Seck, M.; Gorecki, M.; Pescitelli, G.; Desgagne-Penix, I.; Evidente, A. Gigantelline, gigantellinine and gigancrinine, cherylline- and crinine-type alkaloids isolated from Crinum jagus with anti-acetylcholinesterase activity. Phytochemistry 2020, 175, 112390.
- Chen, M.X.; Huo, J.M.; Hu, J.; Xu, Z.P.; Zhang, X. Amaryllidaceae alkaloids from Crinum latifolium with cytotoxic, antimicrobial, antioxidant, and anti-inflammatory activities. Fitoterapia 2018, 130, 48–53.
- Endo, Y.; Sugiura, Y.; Funasaki, M.; Kagechika, H.; Ishibashi, M.; Ohsaki, A. Two new alkaloids from Crinum asiaticum var. japonicum. J. Nat. Med. 2019, 73, 648–652.
- Hanh, T.T.H.; Huong, P.T.T.; Van Thanh, N.; Trung, N.Q.; Van Cuong, T.; Mai, N.T.; Cuong, N.T.; Cuong, N.X.; Nam, N.H.; Van Minh, C. Crinane, augustamine, and β-carboline alkaloids from Crinum latifolium. Phytochem. Lett. 2018, 24, 27–30.
- Cho, N.; Du, Y.; Valenciano, A.L.; Fernandez-Murga, M.L.; Goetz, M.; Clement, J.; Cassera, M.B.; Kingston, D.G.I. Antiplasmodial alkaloids from bulbs of Amaryllis belladonna Steud. Bioorg. Med. Chem. Lett. 2018, 28, 40–42.
- Moodley, N.; Crouch, N.; Bastida, J.; Mulholland, D. Novel alkaloids and a ceramide from Brunsvigia natalensis (Amaryllidaceae) and their anti-neoplastic activity. S. Afr. J. Bot. 2020.
- Katoch, D.; Kumar, D.; Padwad, Y.S.; Singh, B.; Sharma, U. Narciclasine-4-O-beta-d-xylopyranoside, a new narciclasine glycoside from Zephyranthes minuta. Nat. Prod. Res. 2020, 34, 233–240.
- Masi, M.; Frolova, L.V.; Yu, X.; Mathieu, V.; Cimmino, A.; De Carvalho, A.; Kiss, R.; Rogelj, S.; Pertsemlidis, A.; Kornienko, A.; et al. Jonquailine, a new pretazettine-type alkaloid isolated from Narcissus jonquilla quail, with activity against drug-resistant cancer. Fitoterapia 2015, 102, 41–48.
- Presley, C.C.; Krai, P.; Dalal, S.; Su, Q.; Cassera, M.; Goetz, M.; Kingston, D.G.I. New potently bioactive alkaloids from Crinum erubescens. Bioorg. Med. Chem. 2016, 24, 5418–5422.
- Zhan, G.; Qu, X.; Liu, J.; Tong, Q.; Zhou, J.; Sun, B.; Yao, G. Zephycandidine A, the First Naturally Occurring Imidazo[1,2-f]phenanthridine Alkaloid from Zephyranthes candida, Exhibits Significant Anti-tumor and Anti-acetylcholinesterase Activities. Sci. Rep. 2016, 6, 33990.
- Chen, N.; Ji, Y.B.; Zhang, W.G.; Xu, Y.; Yan, X.J.; Sun, Y.F.; Song, H.; Xu, C.R.; Cai, L.P.; Zheng, H.X.; et al. Chemical Constituents from Hymenocallis littoralis. Lett. Org. Chem. 2016, 13, 536–539.
- Brimijoin, S. Molecular forms of acetylcholinesterase in brain, nerve and muscle: Nature, localization and dynamics. Prog. Neurobiol. 1983, 21, 291–322.
- Heller, M.; Hanahan, D.J. Human erythrocyte membrane bound enzyme acetylcholinesterase. Biochim. Biophys. Acta. 1972, 255, 251–272.
- Szelenyi, J.G.; Bartha, E.; Hollan, S.R. Acetylcholinesterase activity of lymphocytes: An enzyme characteristic of T-cells. Br. J. Haematol. 1982, 50, 241–245.
- Darvesh, S.; Hopkins, D.A.; Geula, C. Neurobiology of butyrylcholinesterase. Nat. Rev. Neurosci. 2003, 4, 131–138.
- Lane, R.M.; Potkin, S.G.; Enz, A. Targeting acetylcholinesterase and butyrylcholinesterase in dementia. Int J. Neuropsychopharmacol. 2006, 9, 101–124.
- Sereno, L.; Coma, M.; Rodriguez, M.; Sanchez-Ferrer, P.; Sanchez, M.B.; Gich, I.; Agullo, J.M.; Perez, M.; Avila, J.; Guardia-Laguarta, C.; et al. A novel GSK-3beta inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol. Dis. 2009, 35, 359–367.
- Garcia-Horsman, J.A.; Mannisto, P.T.; Venalainen, J.I. On the role of prolyl oligopeptidase in health and disease. Neuropeptides 2007, 41, 1–24.
- Polgar, L. The prolyl oligopeptidase family. Cell Mol. Life Sci. 2002, 59, 349–362.
- Orhan, I.E. Current concepts on selected plant secondary metabolites with promising inhibitory effects against enzymes linked to Alzheimer’s disease. Curr. Med. Chem. 2012, 19, 2252–2261.
- Babkova, K.; Korabecny, J.; Soukup, O.; Nepovimova, E.; Jun, D.; Kuca, K. Prolyl oligopeptidase and its role in the organism: Attention to the most promising and clinically relevant inhibitors. Future Med. Chem. 2017, 9, 1015–1038.
- Lahiri, D.K.; Farlow, M.R.; Greig, N.H.; Sambamurti, K. Current drug targets for Alzheimer’s disease treatment. Drug Dev. Res. 2002, 56, 267–281.
- Galimberti, D.; Scarpini, E. Old and new acetylcholinesterase inhibitors for Alzheimer’s disease. Expert Opin. Investig. Drugs 2016, 25, 1181–1187.
- López, S.; Bastida, J.; Viladomat, F.; Codina, C. Acetylcholinesterase inhibitory activity of some Amaryllidaceae alkaloids and Narcissus extracts. Life Sci. 2002, 71, 2521–2529.
- Zhan, G.; Zhou, J.; Liu, R.; Liu, T.; Guo, G.; Wang, J.; Xiang, M.; Xue, Y.; Luo, Z.; Zhang, Y.; et al. Galantamine, Plicamine, and Secoplicamine Alkaloids from Zephyranthes candida and Their Anti-acetylcholinesterase and Anti-inflammatory Activities. J. Nat. Prod. 2016, 79, 760–766.
- Vaneckova, N.; Host’alkova, A.; Safratova, M.; Kunes, J.; Hulcova, D.; Hrabinova, M.; Doskocil, I.; Stepankova, S.; Opletal, L.; Novakova, L.; et al. Isolation of Amaryllidaceae alkaloids from Nerine bowdenii W. Watson and their biological activities. Rsc. Adv. 2016, 6, 80114–80120.
- Al Mamun, A.; Maříková, J.; Hulcová, D.; Janoušek, J.; Šafratová, M.; Nováková, L.; Kučera, T.; Hrabinová, M.; Kuneš, J.; Korábečný, J. Amaryllidaceae Alkaloids of Belladine-Type from Narcissus pseudonarcissus cv. Carlton as New Selective Inhibitors of Butyrylcholinesterase. Biomolecules 2020, 10, 800.
- Zhan, G.; Zhou, J.; Liu, J.; Huang, J.; Zhang, H.; Liu, R.; Yao, G. Acetylcholinesterase Inhibitory Alkaloids from the Whole Plants of Zephyranthes carinata. J. Nat. Prod. 2017, 80, 2462–2471.
- Emir, A.; Emir, C.; Bozkurt, B.; Onur, M.A.; Bastida, J.; Somer, N.U. Alkaloids from Galanthus fosteri. Phytochem. Lett. 2016, 17, 167–172.
- Ortiz, J.E.; Pigni, N.B.; Andujar, S.A.; Roitman, G.; Suvire, F.D.; Enriz, R.D.; Tapia, A.; Bastida, J.; Feresin, G.E. Alkaloids from Hippeastrum argentinum and Their Cholinesterase-Inhibitory Activities: An in Vitro and in Silico Study. J. Nat. Prod. 2016, 79, 1241–1248.
- Tallini, L.R.; Torras-Claveria, L.; Borges, W.S.; Kaiser, M.; Viladomat, F.; Zuanazzi, J.A.S.; Bastida, J. N-oxide alkaloids from Crinum amabile (Amaryllidaceae). Molecules 2018, 23, 1277.
- Tallini, L.R.; Osorio, E.H.; Santos, V.D.D.; Borges, W.S.; Kaiser, M.; Viladomat, F.; Zuanazzi, J.A.S.; Bastida, J. Hippeastrum reticulatum (Amaryllidaceae): Alkaloid Profiling, Biological Activities and Molecular Docking. Molecules 2017, 22, 2191.
- Hulcova, D.; Marikova, J.; Korabecny, J.; Hostalkova, A.; Jun, D.; Kunes, J.; Chlebek, J.; Opletal, L.; De Simone, A.; Novakova, L.; et al. Amaryllidaceae alkaloids from Narcissus pseudonarcissus L. cv. Dutch Master as potential drugs in treatment of Alzheimer’s disease. Phytochemistry 2019, 165, 112055.
- Elgorashi, E.E.; Zschocke, S.; van Staden, J. The anti-inflammatory and antibacterial activities of Amaryllidaceae alkaloids. S. Afr. J. Bot. 2003, 69, 448–449.
- Osorio, E.J.; Robledo, S.M.; Bastida, J. Alkaloids with antiprotozoal activity. Alkaloids Chem. Biol. 2008, 66, 113–190.
- Nair, J.J.; van Staden, J. The Amaryllidaceae as a source of antiplasmodial crinane alkaloid constituents. Fitoterapia 2019, 134, 305–313.
- Nair, J.J.; van Staden, J. Antiplasmodial constituents in the minor alkaloid groups of the Amaryllidaceae. S. Afr. J. Bot 2019, 126, 362–370.
- Masi, M.; van der Westhuyzen, A.E.; Tabanca, N.; Evidente, M.; Cimmino, A.; Green, I.R.; Bernier, U.R.; Becnel, J.J.; Bloomquist, J.R.; van Otterlo, W.A.; et al. Sarniensine, a mesembrine-type alkaloid isolated from Nerine sarniensis, an indigenous South African Amaryllidaceae, with larvicidal and adulticidal activities against Aedes aegypti. Fitoterapia 2017, 116, 34–38.
- Masi, M.; Cala, A.; Tabanca, N.; Cimmino, A.; Green, I.R.; Bloomquist, J.R.; van Otterlo, W.A.; Macias, F.A.; Evidente, A. Alkaloids with Activity against the Zika Virus Vector Aedes aegypti (L.)-Crinsarnine and Sarniensinol, Two New Crinine and Mesembrine Type Alkaloids Isolated from the South African Plant Nerine sarniensis. Molecules 2016, 21, 1432.
- Cimmino, A.; Masi, M.; Evidente, M.; Superchi, S.; Evidente, A. Amaryllidaceae alkaloids: Absolute configuration and biological activity. Chirality 2017, 29, 486–499.
- Chaichompoo, W.; Chokchaisiri, R.; Sangkaew, A.; Pabuprapap, W.; Yompakdee, C.; Suksamrarn, A. Alkaloids with anti-human carbonic anhydrase isozyme II activity from the bulbs of Crinum asiaticum L. var. asiaticum. Phytochem. Lett. 2020, 37, 101–105.

