Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Magdalena Klanova | + 4566 word(s) | 4566 | 2021-05-13 04:50:33 | | | |
| 2 | Bruce Ren | -21 word(s) | 4545 | 2021-05-27 03:12:09 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Klanova, M. BCL-2 Proteins in Pathogenesis. Encyclopedia. Available online: https://encyclopedia.pub/entry/10142 (accessed on 08 August 2026).
Klanova M. BCL-2 Proteins in Pathogenesis. Encyclopedia. Available at: https://encyclopedia.pub/entry/10142. Accessed August 08, 2026.
Klanova, Magdalena. "BCL-2 Proteins in Pathogenesis" Encyclopedia, https://encyclopedia.pub/entry/10142 (accessed August 08, 2026).
Klanova, M. (2021, May 26). BCL-2 Proteins in Pathogenesis. In Encyclopedia. https://encyclopedia.pub/entry/10142
Klanova, Magdalena. "BCL-2 Proteins in Pathogenesis." Encyclopedia. Web. 26 May, 2021.
Copy Citation
The ability to inhibit mitochondrial apoptosis is a hallmark of B-cell non-Hodgkin lymphomas (B-NHL). Activation of mitochondrial apoptosis is tightly controlled by members of B-cell leukemia/lymphoma-2 (BCL-2) family proteins via protein-protein interactions. Altering the balance between anti-apoptotic and pro-apoptotic BCL-2 proteins leads to apoptosis evasion and extended survival of malignant cells. The pro-survival BCL-2 proteins: B-cell leukemia/lymphoma-2 (BCL-2/BCL2), myeloid cell leukemia-1 (MCL-1/MCL1) and B-cell lymphoma-extra large (BCL-XL/BCL2L1) are frequently (over)expressed in B-NHL, which plays a crucial role in lymphoma pathogenesis, disease progression, and drug resistance.
apoptosis
non-Hodgkin lymphomas (NHL)
B-cell leukemia/lymphoma-2 (BCL-2)
venetoclax
1. Introduction
Apoptosis is an active form of programmed cell death, in which a controlled sequence of events leads to the elimination of aged, damaged and unnecessary cells [1]. The ability to block apoptosis is a hallmark of many cancers, including hematologic malignancies. Several other types of cell death have been described so far such as necrosis, necroptosis, pyroptosis, ferroptosis, oncosis or autophagy that differ from each other in the triggering process mediating cell death, the involved pathways and/or the impact on the surrounding microenvironment [2]. There are two main apoptotic pathways, the extrinsic or death receptor pathway and the intrinsic or mitochondrial pathway. The extrinsic pathway is triggered by ligation of cell surface death receptors of the tumor necrosis factor (TNF) superfamily, such as TNF receptor 1 or FAS, with specific death ligands [3]. On the contrary, the intrinsic or mitochondrial pathway is triggered by various cellular stresses such as hypoxia, oxidative stress or DNA damage (Figure 1).
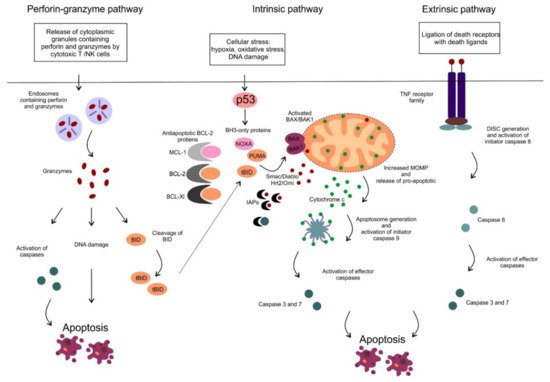
Figure 1. The perforin-granzyme, intrinsic (mitochondrial) and extrinsic apoptotic pathway. The extrinsic pathway is triggered via ligation of cell surface death receptors from the tumor necrosis factor (TNF) receptor family with death ligands (e.g., FAS ligand or TNFα) on the outside of the cell membrane. Subsequently, a multiprotein complex: death-inducing signaling complex (DISC) is created on the cytosolic side of the cell membrane resulting in activation of pro-caspase 8. The initiator caspase 8 subsequently activates the effector caspases (caspase-3 and 7), that cleave cellular proteins and promotes the apoptotic machinery. The perforin-granzyme and intrinsic apoptotic pathways are described in the text.
The two main apoptotic pathways meet at the same execution step resulting in the activation of effector enzymes, cysteine-dependent aspartate-directed proteases, called caspases (Figure 1). Activated caspases possess proteolytic activity and can cleave proteins at aspartic acid residues and target cytoskeletal or nuclear proteins resulting in highly controlled cell disintegration. Another apoptotic pathway has been described, which is mediated by cytotoxic T cells and natural killer (NK) cells (Figure 1). These immune cells can effectively kill infected or transformed cells via the perforin-granzyme apoptotic pathway. Target cells are first exposed to perforin, the protein capable of creating pores in the plasma membrane. Subsequently, secretory granules containing granzyme A and B are released by the immune cells, entering the target cells through the pores. It has also been shown, that perforin and granzymes enter the target cell via receptor-mediated endocytosis (Figure 1) [4]. Granzyme A and B are serine proteases that can cleave proteins at aspartate residues, thus capable of activating caspases and triggering apoptosis [5]. Granzyme B also triggers caspase activation indirectly by activation of pro-apoptotic BH3-only proteins, such as BH3-interacting domain death agonist (BID) which results in the activation of the mitochondrial apoptotic pathway (in detail described below) [6]. In addition, granzyme A can also cleave nuclear proteins and induce single-strand DNA breaks (Figure 1) [7].
Apoptosis is accompanied by characteristic morphological changes of the dying cells. These morphological features are visible via light microscopy and typically include cell shrinkage and nuclear pyknosis caused by chromatin condensation. Another characteristic feature of apoptosis is plasma membrane blebbing with subsequent formation of apoptotic bodies consisting of cytoplasm, intact organelles and nuclear fragments [8]. The apoptotic bodies are cleared via phagocytes without releasing pro-inflammatory cell contents into the surrounding area.
2. Mitochondrial Apoptotic Pathway
In this review, we focus on the mitochondrial apoptotic pathway, which plays a critical role in pathogenesis and drug resistance of distinct hematologic malignancies and has been extensively studied and targeted therapeutically. The activation of the mitochondrial pathway leads to increased mitochondrial outer membrane permeability (MOMP), loss of mitochondrial transmembrane potential and oxidative phosphorylation and release of multiple pro-apoptotic molecules such as second mitochondrial-derived activator of caspases/direct inhibitor of apoptosis binding protein with low pI (Smac/DIABLO), the serine protease HtrA2/Omi and cytochrome c. Smac/DIABLO, as well as HtrA2/Omi, promote apoptosis via interaction with inhibitor of apoptosis proteins (IAPs) [9][10]. IAPs can interact with caspases, inhibit their function and protect cells from apoptosis [11]. Cytochrome c is normally sequestered within the mitochondrial intermembrane space playing an essential role as an electron carrier in the respiratory chain. Once released from mitochondria, cytochrome c associates with apoptotic protease-activating factor 1 (APAF-1), deoxyadenosine triphosphate and procaspase 9, assembling a multiprotein complex called apoptosome. In the apoptosome the initiator procaspase 9 is processed and activated into caspase 9, harboring the proteolytic activity to cleave the effector procaspase 3. Activated caspase 3 subsequently cleaves cellular proteins and promotes the apoptotic machinery (Figure 1) [12].
The permeability of the outer mitochondrial membrane is tightly controlled by members of BCL-2 family proteins via protein-protein interactions. The BCL-2 family of proteins consists of more than 20 different members classified based on the protein structure and key functions into three main subgroups, the anti-apoptotic proteins, the pro-apoptotic multi-domain effector proteins, and pro-apoptotic BCL-2 homology (BH)3 domain-only proteins [13]. Altering the balance between anti-apoptotic and pro-apoptotic BCL-2 proteins can lead to disruption of the apoptotic process and the aberrant survival of cancer cells [14]. The pro-survival BCL-2 proteins comprise B-cell leukemia/lymphoma-2 (BCL-2), myeloid cell leukemia-1 (MCL-1), B-cell lymphoma-extra large (BCL-XL; BCL2L1), B-cell lymphoma-w (BCL-W/BCL2L2), BCL2-like protein 10 (BCL-B/BCL2L10) and BCL-2 related gene A1 (A1/BCL2A1; also known as BFL-1). The pro-survival BCL-2 proteins sequester the pro-apoptotic BCL-2 proteins thereby mediating their anti-apoptotic activity. The anti-apoptotic BCL-2 proteins share a common structure with a transmembrane domain, which is necessary for anchoring to cellular membranes, and four conserved motifs called BH domains (BH1, BH2, BH3, BH4). BH1, BH2, and BH3 domains create a hydrophobic pocket, which is critical for interaction with pro-apoptotic BCL-2 proteins. Interestingly, some proteins of this group have isoforms translated from the same gene that possess pro-apoptotic activity. For example, there are three known isoforms of MCL-1 including full-length MCL-1 (MCL-1L), MCL-1 short (MCL-1S) and MCL-1 extra short (MCL-1ES) generated by alternative splicing of the same gene. In contrast to the anti-apoptotic protein MCL-1L, both MCL-1S and MCL-1ES have different protein structures and display pro-apoptotic activities [15].
Based on their protein structure and key functions, the pro-apoptotic proteins of the BCL-2 family can be divided into following groups: (1) multi-domain effector proteins including BCL-2–associated X protein (BAX), BCL-2 antagonist/killer 1 (BAK1) and BCL-2 homologous antagonist killer (BOK), and (2) BH3 domain-only proteins that comprise BCL-2-interacting mediator of cell death (BIM/BCL2L11), BID, NOXA/PMAIP1, BCL-2 antagonist of cell death (BAD), p53 upregulated modulator of apoptosis (PUMA/BBC3) and harakiri (HRK).
The multi-domain effector proteins BAX, BAK1, and BOK contain a transmembrane domain and three BH3 domains (BH1, BH2, BH3). Activated proteins BAX and BAK form homodimers and heterodimers and create pore-like structures in the outer mitochondrial membrane resulting in its increased permeability for intermembrane space molecules [16]. The precise function of the third member of this family, BOK remains elusive [17]. The so-called BH3 domain-only pro-apoptotic proteins contain only one BH3 domain and some of them also one transmembrane domain. Two models of apoptosis induction have been proposed. In the direct activation model, the BH3 domain-only proteins act either as direct activators of apoptosis (BIM, BID, and perhaps PUMA), possessing the ability to activate the effectors BAX and BAK1, or as sensitizers (NOXA, BAD, HRK). Because the sensitizers cannot directly interact with BAX or BAK1, their pro-apoptotic effect is mediated through binding to anti-apoptotic BCL-2 proteins and subsequent release of BH3 domain-only activators. In the second, indirect model of apoptosis, permanently active forms of BAX and BAK1 are bound to the anti-apoptotic BCL-2 proteins. Increased expression of pro-apoptotic proteins leads to the displacement of these active forms of pro-apoptotic effector proteins, which triggers the mitochondrial apoptosis. Selective binding and different affinities between anti-apoptotic and BH3 domain-only proteins have been shown. While BH3 domain-only activators bind to all anti-apoptotic proteins, BH3 domain-only sensitizers display selective interactions with anti-apoptotic proteins (NOXA-MCL-1, HRK-BCL-XL) caused by differences in the amino acid sequence of the BH3 domain [18] (Figure 2). Importantly, it has been repeatedly demonstrated that cancer cells, whose anti-apoptotic BCL-2 proteins are occupied by BH3 domain-only activators are so-called “primed for death” [18]. Such cells can undergo rapid apoptosis, when exposed to BH3 domain-only sensitizers or BH3 mimetics, a class of anti-tumor molecules that displace BH3 domain-only activators to activate BAX/BAK1 and trigger apoptosis [19].
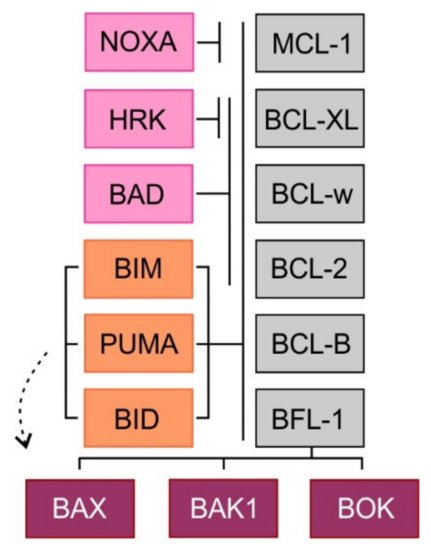
Figure 2. Binding profile of pro-apoptotic BH3 domain-only proteins and multi-domain effector proteins to pro-survival proteins. BH3 domain-only proteins are depicted in orange (direct activators) and pink (sensitizers).
3. B-Cell Non-Hodgkin Lymphomas: Pathogenesis and Classification
B-cell non-Hodgkin lymphomas (B-NHLs) represent a heterogeneous group of hematologic malignancies, each arising from different non-malignant lymphoid counterparts (Figure 3) [20]. B cells develop from hematopoietic stem cells in the bone marrow compartment, the primary lymphoid organ and upon their release into peripheral blood, B cells mature in secondary lymphoid tissues including lymph nodes, spleen or tonsils. B cell development comprises different stages, during which lymphocytes undergo critical processes necessary for their proper development, including generation and expression of a functional surface B-cell receptor (BCR). One of the first events to occur is VDJ recombination, the process in which double-strand DNA breaks are randomly introduced by recombinase activating gene RAG1 and RAG2 into gene segments encoding variable (V), diversity (D) and joining (J) regions of the BCR with following DNA repair by non-homologous end joining [21]. This process ensures high variability of BCRs on the surface of B-cells capable to face multiple antigens during the immune response [22]. Once the surface BCR is expressed, B cells leave the bone marrow, becoming mature naïve B cells ready to be exposed to various antigens. Another two events modifying the coding sequence of BCR occur in secondary lymphoid tissues: somatic hypermutation (SHM) and class switch recombination (CSR). Both events are mediated by activation-induced cytidine deaminase (AID) [23]. In the case of SHM, AID introduces random mutations into the coding sequence of the variable region of the BCR, which results in a changed affinity for the immunizing antigens. While a randomly increased affinity to antigen would foster the pro-survival signaling from BCR and increase the mitotic activity of the lymphocyte, a decreased affinity would lead to triggering apoptosis and demise of the lymphocyte clone. CSR that enables the switching of the heavy chain class of Ig molecule (e.g., from IgM to IgG) is implemented by DNA recombination. Unfortunately, VDJ recombination, SHM, and CSR are prone to mistakes that can introduce genetic alterations of the developing lymphocytes and contribute to their malignant transformation (Figure 3) [20].
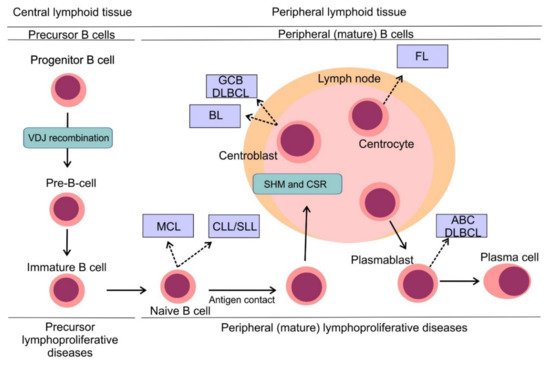
Figure 3. Pathogenesis of B-cell non-Hodgkin lymphomas. Simplified scheme of B cell development showing distinct types of B-NHLs arising from different non-malignant lymphoid counterparts. Reprinted with permission. © (2020) American Society of Clinical Oncology. All rights reserved. Nogai, H. et al.: J. Clin. Oncol. 29, 2011: 1803–1811 [20].
The recent World Health Organization (WHO) classification of lymphoid malignancies identifies approximately fifty mature lymphoproliferative disorders of B-cell origin with distinct clinical, pathological and genetic features [24]. Lymphomas can be divided into aggressive (high-mitotic activity) and indolent (low-mitotic activity) subtypes, which reflects the clinical behavior of these entities. Aggressive lymphomas require immediate treatment, while indolent lymphomas can be subject to watchful waiting in a large proportion of patients. Diffuse large B-cell lymphoma (DLBCL) represents the most common lymphoma subtype and accounts for 30%–40% cases in adults [25]. DLBCL is an aggressive lymphoma subtype requiring treatment upon diagnosis. Two, histologically indistinguishable DLBCL subtypes have been identified by gene expression profiling, each arising from a different cell of origin (COO) [26]. Germinal center B-cell-like (GCB) and activated B-cell-like COO DLBCL subtypes are each driven by distinct oncogenic pathways, display different clinical behavior and have different clinical outcomes, with ABC DLBCL having significantly worse outcome compared to GCB DLBCL [27][28]. Follicular lymphoma (FL) is the second most prevalent subtype of malignant lymphomas and accounts for approximately 20% of all lymphoma cases in adults [25]. It is typically an indolent disease with long term survival. Other frequently diagnosed aggressive B-NHL include mantle cell lymphoma (MCL) and Burkitt lymphoma (BL), while other prevalent indolent lymphomas comprise marginal zone lymphoma (MZL) and small lymphocytic lymphoma (SLL). On a molecular level, SLL refers to the same disease as chronic lymphocytic leukemia (CLL) with specific differences in the clinical picture. CLL is the most common leukemia of the adult in the Western hemisphere but is a rare disease in the Far East. CLL typically presents with hyperlymphocytosis, while the dominant finding in the clinical picture of patients with SLL is lymph node involvement.
4. Deregulation of BCL-2 Proteins in B-Cell Non-Hodgkin Lymphomas
Deregulation of mitochondrial apoptosis is a hallmark of lymphomas. Indeed, BCL-2 was originally discovered because of its involvement in translocation t(14;18) in follicular lymphoma [29]. Later on, it was discovered that the aberrant expression of BCL-2 protein contributes to the pathogenesis of many types of human malignancies, including leukemias, lymphomas, and cancers. Within B-NHL BCL-2 overexpression commonly arises from genetic abnormalities. Importantly, the frequency of these alterations, as well as the extent of BCL2 mRNA and protein expressions, are substantially different in distinct lymphoma subtypes (Table 1).
Table 1. Mechanisms of B-cell leukemia/lymphoma-2 (BCL-2) overexpression in B-cell non-Hodgkin lymphomas (B-NHL).
| B-NHL Subtype | BCL-2 Positivity * | Mechanism of BCL-2 Overexpression |
|---|---|---|
| DLBCL | 49%–67% [30][31][32] | BCL2 translocation in GCB DLBCL (30% of cases) [33] BCL2 amplification in ABC DLBCL (20% of cases) [34] |
| FL | > 90% [35][36][37] | BCL2 translocation (90% of cases) [35][36] |
| MCL | BCL-2 positive [38] | 13q14.3 loss (55% of cases) [39] 18q21 gains (20% of cases) [37][39] |
| BL | BCL-2 negative, weak expression in up to 20% [40] | NA |
| CLL/SLL | BCL-2 positive (high expression in majority of the cases) [41] | 13q14.3 loss (68% cases) [42] BCL2 gene hypomethylation [43] BCL2 translocation (rare, < 5%) [41] |
| MZL | >80% [44] | unknown |
Chromosomal translocation t(14;18)(q32;q21), juxtaposing BCL2 gene (18q) under the control of the immunoglobulin heavy chain gene promoter (14q), leads to constitutive expression of BCL-2, inhibition of apoptosis and extended cell survival [45]. Translocation t(14;18)(q32;q21) is a genetic hallmark of follicular lymphoma (FL) and is detected in approximately 90% of all FL cases. The remainder 10% FL cases lack BCL2 gene translocation and display distinct molecular features with activated B cell-like, NFκB and proliferation expression profiles and frequent lack of BCL-2 protein expression. Interestingly, no differences in overall survival have been shown between translocation-positive and negative FL cases [35]. Genetic alterations (chromosomal translocations, gene amplification, and single nucleotide variants) of BCL2 genes are frequent abnormalities in DLBCL, however, the frequency of distinct alterations, as well as prevalence of BCL-2 positivity differ between the two major COO subtypes. The translocations t(14;18)(q32;q21), detected in more than 30% GCB DLBCL have been associated with high BCL-2 protein expression and poor outcome [33]. Interestingly, this chromosomal aberration is not detected in ABC DLBCL [33][46]. Amplifications of 18q21 locus resulting in BCL-2 overexpression are in contrary significantly more prevalent in ABC DLBCL and are detected in approximately 20% of ABC DLBCL cases [34]. BCL2 SNVs can be found in approximately 8% of all DLBCL cases and are more frequently detected in the GCB than in the ABC DLBCL subtype [33]. BCL2 SNVs usually co-occur with BCL2 translocation, possibly as a consequence of ongoing aberrant somatic hypermutation [47]. SNVs tend to be located in the flexible loop domain of BCL2 gene, while mutations in BH domains that could impact interaction with BH3 mimetics are rare [47]. Some studies showed that BCL2 SNVs were associated with shorter progression-free survival while other studies did not [33][47]. In mantle cell lymphoma (MCL), BCL-2 protein is overexpressed in virtually all cases. Similarly to ABC DLBCL, BCL2 amplification is frequently found in MCL, while the translocations are rare [39]. Another cytogenetic abnormality contributing to high BCL-2 protein expression in MCL is loss of 13q14 locus by deletion [48]. The cluster at 13q14.3 contains genes for two microRNAs, miR-15a and miR-16-1, both of which negatively regulate BCL2 at the posttranscriptional level. The loss of this chromosomal region thus results in high BCL-2 expression [49]. Similarly, high BCL-2 protein expression can be documented in virtually all patients with CLL and deletion of 13q14 is common in CLL [43][50][51]. Another mechanism contributing to high BCL-2 expression in CLL is hypomethylation of BCL2 gene [43]. In contrast to the above mentioned B-NHLs, the level of BCL-2 expression in Burkitt lymphoma is low or undetectable, which has been used as a part of the diagnostic algorithm of this lymphoma subtype [40].
Although the role of other anti-apoptotic proteins in the pathogenesis of B-NHL is less clear, it is probable that various B-NHL subtypes rely on more than one anti-apoptotic protein [52]. The potential role of MCL-1 in lymphoma pathogenesis was demonstrated in transgenic mouse models in which MCL-1 transgenic mice developed B-cell lymphomas at high frequency [53]. Limited data show that MCL-1 protein is highly expressed in aggressive B-NHL, including DLBCL (84% cases), BL (89% cases) and in grade 3 FL (100%). Other B-NHL subtypes (MCL, MZL, SLL) display lower and less frequent MCL-1 positivity [54][55]. Molecular mechanisms leading to aberrant MCL-1 expression have been studied in DLBCL. It has been shown that high MCL-1 expression is more frequent in the ABC subtype. MCL1 locus (1q21) gain/amplification and constitutive activation of the STAT3 pathway were identified as key drivers of aberrant MCL-1 expression in this lymphoma subtype [56].
Although BCL-XL appears to be frequently expressed, its overexpression responsible for resistance to specific BCL-2 inhibitors has been observed in response to pro-survival signaling from the lymph node microenvironment rather than genetic alterations. Moreover, BCL-XL is critical for the proper development of platelets, and targeted inhibition of BCL-XL has been associated with thrombocytopenia (see Section 5.). Another anti-apoptotic protein, BCL-W has been shown to be overexpressed in the majority of aggressive and indolent lymphoma, including DLBCL (equally in both COO subtypes), BL, MCL, FL, and MZL, making it a potential therapeutic target in B-NHL [52].
Although overexpression of pro-survival BCL-2 proteins as a consequence of genetic alterations is a frequent event in B-NHL, the delicate balance between anti-apoptotic and pro-apoptotic BCL-2 proteins might be disrupted by lack of pro-apoptotic BCL-2 proteins, leading to apoptosis evasion and survival advantage. The key role of pro-apoptotic protein BIM in the pathogenesis of MCL has been proposed in a transgenic mouse model, when cyclin D1-transgenic mice harboring BIM-deficient B cells developed lymphomas with histopathologic and molecular features of human MCL [57]. Although approximately one-third of currently available MCL cell lines harbor mono- or biallelic BIM/BCL2L11 deletion, we have shown that this alteration is rarely found in patients with newly diagnosed MCL [58]. Even though a complete lack of BIM protein expression as a result of gene deletion is probably rare, the level of BIM protein expression was shown to negatively correlate with prognosis in MCL [59].
Besides frequent alterations of genes encoding BCL-2 proteins, lymphomas may evade apoptosis through activated signaling pathways, resulting in transcriptional deregulation of BCL-2 proteins. Deregulation of the phosphate and tensin homolog (PTEN)-phosphatidylinositol 3-kinase (PI3K) pathway plays an important role in the pathogenesis of GCB DLBCL. Loss of PTEN, either by gene deletion or amplification of miR-17-92, which suppresses PTEN, can be found in approximately 55% GCB DLBCL [60]. Loss of PTEN leads to activation of PI3K-AKT pathway and upregulation of downstream targets, including e.g., transcription factor MYC, thus promoting growth, survival, and proliferation [60]. It has been shown that Akt-mediated phosphorylation inhibits the pro-apoptotic protein BAD. Akt phosphorylates BAD at serine 112 resulting in its dissociation from BCL-2 to form a complex with the 14-3-3 adaptor protein, which is associated with cell survival [61]. On the contrary, constitutive activation of NFκB pathway is a hallmark of ABC DLBCL and several studies have shown the dependence of ABC DLBCL on NFκB signaling [62][63]. NFκB is a family of related transcription factors that are normally kept inactive in the cytoplasm by interaction with specific inhibitors. Stimulation through various receptors, including the surface B-cell receptor, results in the release of NFκB factors, their activation, translocation to the nucleus and transcription of target genes. Three upstream molecules CARD11, BCL-10/BCL10 and MALT-1, forming a CBM complex, are necessary to convert signals from the BCR into the cell to activate the so-called canonical NFκB pathway [63]. Missense mutations of CARD11, leading to strong activation of NFκB signaling, are frequent events in both GCB and ABC DLBCL [33][64]. Another pathogenic mutation identified a few years later was a gain of function mutation of MYD88, an adaptor protein that mediates toll and interleukin (IL)-1 receptor signaling and activates NFκB pathway in distinct lymphoma subtypes. MYD88 (L265P) can only be detected in ABC DLBCL and in a subset of MZL called mucosa-associated lymphoid tissue (MALT) lymphoma [65]. Other well-defined molecular events deregulating the CBM complex have been reported in MALT lymphomas. Chromosomal translocations t(14;18)(q32;q21) and t(1;14)(p22;q32), bringing MALT1 and BCL10 under the control of the immunoglobulin heavy chain promoter have been associated with this lymphoma subtype. Both MALT1 and BCL10 translocations can make CBM complex formation independent of upstream signaling, resulting in constitutive NFκB pathway activation [66][67]. The third translocation involved in aberrant NFκB signaling in this lymphoma subtype is the t(11;18)(q21;q21), resulting in the expression of a chimeric IAP2-MALT-1 protein. IAP2-MALT-1 can interact and deregulate components of a multiprotein IκB-kinase (IKK) complex, resulting in NFκB pathway activation [68]. Known NFκB targets that promote cell survival via inhibition of apoptosis include anti-apoptotic BCL-2 proteins BCL-XL and A1, caspase inhibitors from the IAP family and Cellular FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein (c-FLIP) [69].
Another important mechanism of how lymphomas evade apoptosis is the deregulation of the DNA damage pathway. TP53 alterations (deletions, mutations) are recurrently found in aggressive lymphomas but are rather rare in indolent lymphomas [70]. Prognostically, TP53 aberrations have repeatedly correlated with adverse outcome and chemoresistance [71][72][73]. In indolent lymphomas, TP53 aberrations have been associated with the transformation to aggressive lymphomas [74][75]. P53 directly transactivates pro-apoptotic BH3-only proteins PUMA and NOXA [76][77]. As a consequence of structural or functional p53 inactivation, the critical DNA damage response is disrupted, which increases genomic instability and facilitates the survival of lymphoma cells in response to genotoxic cytostatics.
5. Therapeutic Inhibition of Anti-Apoptotic BCL-2 Proteins
Given the fact that anti-apoptotic BCL-2 proteins play a crucial role in lymphoma pathogenesis, disease progression and drug resistance, the efforts to target them therapeutically have been underway for several decades. Moreover, therapeutic targeting of BCL-2 proteins is upstream and, therefore, independent of TP53, the key tumor suppressor gene frequently mutated in distinct lymphoid malignancies, overcoming the negative prognostic impact of this genetic alteration. Due to its indisputable role in the pathogenesis of B-NHL, BCL-2 protein has been in the spotlight of extensive efforts to develop targeted therapies. Several approaches have been explored to target this protein, including antisense oligonucleotides inhibiting expression of BCL-2 protein, peptide inhibitors or small molecule inhibitors.
Oblimersen sodium (G3139, Genansense, Genta Inc, Berkeley Heights, NJ, USA) was the first BCL-2 inhibitor tested in clinical practice. Oblimersen sodium is a phosphorothioated 18 basis DNA molecule, which was designed to complementary bind to the first six codons of BCL2 mRNA leading to BCL2 mRNA degradation and decrease in BCL-2 protein expression [78]. Despite evidence of efficacy in early clinical trials in hematological malignancies including CLL, the phase III trial combining oblimersen sodium with (immuno-) chemotherapy in patients with relapsed or refractory CLL failed to show clear clinical benefit and oblimersen sodium has never been approved by the US Food and Drug Administration (FDA) [79].
Another attempt to directly target anti-apoptotic BCL-2 proteins was made with small molecule inhibitor, obatoclax (GX-15-070). Obatoclax binds to all key anti-apoptotic proteins, BCL-2, BCL-XL and MCL-1 resulting in the displacement of pro-apoptotic proteins BAX, BAK or BIM and apoptosis induction [80]. Other mechanisms of action of obatoclax have been described including S/G2 cell cycle arrest resulting in growth inhibition. These observations indicate that besides anti-apoptotic BCL-2 proteins obatoclax has other targets [81]. Obatoclax has been tested in several phase 1-2 clinical trials in various types of B-NHL and CLL. These trials showed modest clinical activity of obatoclax as a single agent and did not confirm the synergistic effect of obatoclax with various anti-lymphoma agents suggested in preclinical studies. Moreover, neurological toxicity has been observed in all these trials [82][83][84][85]. Both, the modest clinical activity as well as the toxicity profile limited development of obatoclax as a therapeutic agent in B/NHL and CLL.
A breakthrough in the development of BCL-2 inhibitors was achieved with ABT-737, a small molecule inhibitor of BCL-2, BCL-XL, and BCL-W. ABT-737 displays a much higher binding affinity to anti-apoptotic proteins than obatoclax, with subsequent release of proapoptotic proteins and BAX/BAK1-dependent apoptosis induction [86]. ABT-263 (Navitoclax, Abbott Laboratories, North Chicago, IL, USA) is a chemically closely related agent with a similar binding affinity to BCL-2 proteins, which is orally available [87]. The promising preclinical activity led to the clinical testing of Navitoclax in various B-NHL and CLL. Phase 1 study of navitoclax in patients with relapsed or refractory lymphoid malignancies demonstrated clinical activity across all histological subtypes with the best activity seen in CLL [88]. A follow-up phase 1 dose-escalation study of single-agent navitoclax in patients with relapsed or refractory CLL confirmed the efficacy of navitoclax with durable responses and median progression-free survival of 25 months in heavily pretreated CLL patients [89]. The dose-limiting toxicity of navitoclax observed in these trials was thrombocytopenia. Rapid reduction in platelet count (grade 3–5) was observed in 28% of patients and prevented the exploration of higher doses of navitoclax [89]. Thrombocytopenia was caused by inhibition of BCL-XL in platelets, that was shown to be dependent on BCL-XL for survival [90][91]. Further phase 2 trial of navitoclax in combination with rituximab vs rituximab alone in patients with previously untreated CLL demonstrated improved objective response rate (ORR) of rituximab plus navitoclax vs rituximab alone (50% vs 35%, respectively), with a further increase of ORR (70%) when navitoclax with rituximab was administered until progression [92]. It has been shown that CLL patients with del(17p) or high levels of BCL-2 had significantly better clinical responses when treated with navitoclax [92]. This observation together with the navitoclax-induced thrombocytopenia caused by BCL-XL inhibition led to efforts to develop a BCL-2 specific inhibitor.
Venetoclax (ABT-199/GDC-0199) was the first highly selective orally available BCL-2 inhibitor developed by the re-engineering of navitoclax [93]. Similarly to navitoclax, venetoclax displays high affinity for BCL-2 (Ki < 0.010 nM), however its binding affinity to other anti-apoptotic proteins including BCL-XL (Ki = 48 nM), MCL-1 (Ki > 444 nM) and BCL-W (Ki = 245 nM) is much weaker [93]. The mode of action of venetoclax is shown in Figure 4A. Preclinical in vitro and in vivo studies in various human hematological cell lines and xenograft models demonstrated significant anti-tumor efficacy which led to early clinical testing [93].
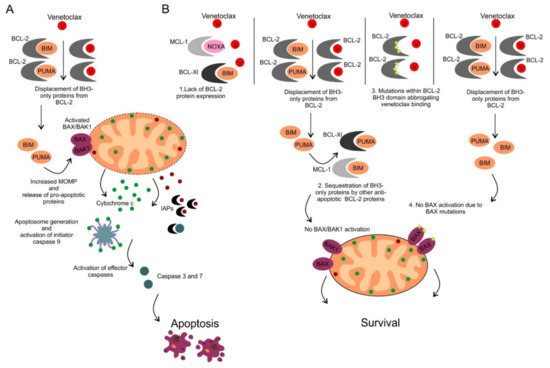
Figure 4. (A) Mechanism of action of venetoclax. Venetoclax binds to the BH3 domain of BCL-2 protein with subsequent release of proapoptotic proteins, activation of BAX/BAK1 and apoptosis induction. (B) Mechanisms of resistance to venetoclax. Four different mechanisms of inherited or acquired resistance to venetoclax include: 1. Lack of BCL-2 protein expression, 2. Overexpression of other anti-apoptotic BCL-2 proteins that sequester pro-apoptotic BH3-only proteins displaced from BCL-2 protein following its pharmacological inhibition by venetoclax, 3. Acquired mutations within the BH3 domain of BCL-2 abrogating venetoclax binding, 4. Acquired mutations of BAX preventing BAX activation and apoptosis induction.
References
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257.
- Dixon, S.J. Ferroptosis: Bug or feature? Immunol. Rev. 2017, 277, 150–157.
- Ashkenazi, A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat. Rev. Cancer 2002, 2, 420–430.
- Trapani, J.A.; Smyth, M.J. Functional significance of the perforin/granzyme cell death pathway. Nat. Rev. Immunol. 2002, 2, 735–747.
- Voskoboinik, I.; Whisstock, J.C.; Trapani, J.A. Perforin and granzymes: Function, dysfunction and human pathology. Nat. Rev. Immunol. 2015, 15, 388–400.
- Sutton, V.R.; Davis, J.E.; Cancilla, M.; Johnstone, R.W.; Ruefli, A.A.; Sedelies, K.; Browne, K.A.; Trapani, J.A. Initiation of apoptosis by granzyme B requires direct cleavage of bid, but not direct granzyme B-mediated caspase activation. J. Exp. Med. 2000, 192, 1403–1414.
- Beresford, P.J.; Xia, Z.; Greenberg, A.H.; Lieberman, J. Granzyme A loading induces rapid cytolysis and a novel form of DNA damage independently of caspase activation. Immunity 1999, 10, 585–594.
- Huppertz, B.; Frank, H.G.; Kaufmann, P. The apoptosis cascade--morphological and immunohistochemical methods for its visualization. Anat. Embryol. (Berl) 1999, 200, 1–18.
- Hegde, R.; Srinivasula, S.M.; Zhang, Z.; Wassell, R.; Mukattash, R.; Cilenti, L.; DuBois, G.; Lazebnik, Y.; Zervos, A.S.; Fernandes-Alnemri, T.; et al. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J. Biol. Chem. 2002, 277, 432–438.
- Yang, Q.H.; Church-Hajduk, R.; Ren, J.; Newton, M.L.; Du, C. Omi/HtrA2 catalytic cleavage of inhibitor of apoptosis (IAP) irreversibly inactivates IAPs and facilitates caspase activity in apoptosis. Genes Dev. 2003, 17, 1487–1496.
- Liu, Z.; Sun, C.; Olejniczak, E.T.; Meadows, R.P.; Betz, S.F.; Oost, T.; Herrmann, J.; Wu, J.C.; Fesik, S.W. Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature 2000, 408, 1004–1008.
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193.
- Warren, C.F.A.; Wong-Brown, M.W.; Bowden, N.A. BCL-2 family isoforms in apoptosis and cancer. Cell Death Dis. 2019, 10, 177.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Kim, J.H.; Bae, J. MCL-1ES induces MCL-1L-dependent BAX- and BAK-independent mitochondrial apoptosis. PLoS ONE 2013, 8, e79626.
- Nechushtan, A.; Smith, C.L.; Lamensdorf, I.; Yoon, S.H.; Youle, R.J. Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J. Cell Biol. 2001, 153, 1265–1276.
- Ke, F.; Bouillet, P.; Kaufmann, T.; Strasser, A.; Kerr, J.; Voss, A.K. Consequences of the combined loss of BOK and BAK or BOK and BAX. Cell Death Dis. 2013, 4, e650.
- Certo, M.; Del Gaizo Moore, V.; Nishino, M.; Wei, G.; Korsmeyer, S.; Armstrong, S.A.; Letai, A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006, 9, 351–365.
- Merino, D.; Kelly, G.L.; Lessene, G.; Wei, A.H.; Roberts, A.W.; Strasser, A. BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 2018, 34, 879–891.
- Nogai, H.; Dorken, B.; Lenz, G. Pathogenesis of non-Hodgkin’s lymphoma. J. Clin. Oncol. 2011, 29, 1803–1811.
- Schatz, D.G.; Oettinger, M.A.; Baltimore, D. The V(D)J recombination activating gene, RAG-1. Cell 1989, 59, 1035–1048.
- LeBien, T.W.; Tedder, T.F. B lymphocytes: How they develop and function. Blood 2008, 112, 1570–1580.
- Muramatsu, M.; Kinoshita, K.; Fagarasan, S.; Yamada, S.; Shinkai, Y.; Honjo, T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000, 102, 553–563.
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390.
- The Non-Hodgkin’s Lymphoma Classification Project. A Clinical Evaluation of the International Lymphoma Study Group Classification of Non-Hodgkin’s Lymphoma. Blood 1997, 89, 3909–3918.
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511.
- Vitolo, U.; Trneny, M.; Belada, D.; Burke, J.M.; Carella, A.M.; Chua, N.; Abrisqueta, P.; Demeter, J.; Flinn, I.; Hong, X.; et al. Obinutuzumab or Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone in Previously Untreated Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2017.
- Lenz, G.; Wright, G.; Dave, S.S.; Xiao, W.; Powell, J.; Zhao, H.; Xu, W.; Tan, B.; Goldschmidt, N.; Iqbal, J.; et al. Stromal gene signatures in large-B-cell lymphomas. N. Engl. J. Med. 2008, 359, 2313–2323.
- Tsujimoto, Y.; Finger, L.R.; Yunis, J.; Nowell, P.C.; Croce, C.M. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 1984, 226, 1097–1099.
- Scott, D.W.; Mottok, A.; Ennishi, D.; Wright, G.W.; Farinha, P.; Ben-Neriah, S.; Kridel, R.; Barry, G.S.; Hother, C.; Abrisqueta, P.; et al. Prognostic Significance of Diffuse Large B-Cell Lymphoma Cell of Origin Determined by Digital Gene Expression in Formalin-Fixed Paraffin-Embedded Tissue Biopsies. J. Clin. Oncol. 2015, 33, 2848–2856.
- Hu, S.; Xu-Monette, Z.Y.; Tzankov, A.; Green, T.; Wu, L.; Balasubramanyam, A.; Liu, W.M.; Visco, C.; Li, Y.; Miranda, R.N.; et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: A report from The International DLBCL Rituximab-CHOP Consortium Program. Blood 2013, 121, 4021–4031.
- Sehn, L.H.; Oestergaard, M.Z.; Trněný, M.; Bosi, A.; Egyed, M.; Illes, A.; Nakamae, H.; Opat, S.; Topp, M.; Zaja, F.; et al. Prognostic impact of bcl2 and myc expression and translocation in untreated dlbcl: Results from the phase iii goya study. Hematol. Oncol. 2017, 35, 131–133.
- Bolen, C.R.; Klanova, M.; Trneny, M.; Sehn, L.H.; He, J.; Tong, J.; Paulson, J.N.; Kim, E.; Vitolo, U.; Di Rocco, A.; et al. Prognostic impact of somatic mutations in diffuse large B-cell lymphoma and relationship to cell-of-origin: Data from the phase III GOYA study. Haematologica 2019.
- Iqbal, J.; Neppalli, V.T.; Wright, G.; Dave, B.J.; Horsman, D.E.; Rosenwald, A.; Lynch, J.; Hans, C.P.; Weisenburger, D.D.; Greiner, T.C.; et al. BCL2 expression is a prognostic marker for the activated B-cell-like type of diffuse large B-cell lymphoma. J. Clin. Oncol. 2006, 24, 961–968.
- Leich, E.; Salaverria, I.; Bea, S.; Zettl, A.; Wright, G.; Moreno, V.; Gascoyne, R.D.; Chan, W.C.; Braziel, R.M.; Rimsza, L.M.; et al. Follicular lymphomas with and without translocation t(14;18) differ in gene expression profiles and genetic alterations. Blood 2009, 114, 826–834.
- Bende, R.J.; Smit, L.A.; van Noesel, C.J. Molecular pathways in follicular lymphoma. Leukemia 2007, 21, 18–29.
- Guo, Y.; Karube, K.; Kawano, R.; Suzumiya, J.; Takeshita, M.; Kikuchi, M.; Huang, G.-S.; Li, Q.; Ohshima, K. Bcl2-negative follicular lymphomas frequently have Bcl6 translocation and/or Bcl6 or p53 expression. Pathol. Int. 2007, 57, 148–152.
- Davids, M.S.; Roberts, A.W.; Seymour, J.F.; Pagel, J.M.; Kahl, B.S.; Wierda, W.G.; Puvvada, S.; Kipps, T.J.; Anderson, M.A.; Salem, A.H.; et al. Phase I First-in-Human Study of Venetoclax in Patients With Relapsed or Refractory Non-Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 826–833.
- Tagawa, H.; Karnan, S.; Suzuki, R.; Matsuo, K.; Zhang, X.; Ota, A.; Morishima, Y.; Nakamura, S.; Seto, M. Genome-wide array-based CGH for mantle cell lymphoma: Identification of homozygous deletions of the proapoptotic gene BIM. Oncogene 2005, 24, 1348–1358.
- Masque-Soler, N.; Szczepanowski, M.; Kohler, C.W.; Aukema, S.M.; Nagel, I.; Richter, J.; Siebert, R.; Spang, R.; Burkhardt, B.; Klapper, W. Clinical and pathological features of Burkitt lymphoma showing expression of BCL2--an analysis including gene expression in formalin-fixed paraffin-embedded tissue. Br. J. Haematol. 2015, 171, 501–508.
- Majid, A.; Tsoulakis, O.; Walewska, R.; Gesk, S.; Siebert, R.; Kennedy, D.B.; Dyer, M.J. BCL2 expression in chronic lymphocytic leukemia: Lack of association with the BCL2 938A>C promoter single nucleotide polymorphism. Blood 2008, 111, 874–877.
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529.
- Hanada, M.; Delia, D.; Aiello, A.; Stadtmauer, E.; Reed, J.C. bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia. Blood 1993, 82, 1820–1828.
- Lai, R.; Arber, D.A.; Chang, K.L.; Wilson, C.S.; Weiss, L.M. Frequency of bcl-2 expression in non-Hodgkin’s lymphoma: A study of 778 cases with comparison of marginal zone lymphoma and monocytoid B-cell hyperplasia. Mod. Pathol. 1998, 11, 864–869.
- Bakhshi, A.; Jensen, J.P.; Goldman, P.; Wright, J.J.; McBride, O.W.; Epstein, A.L.; Korsmeyer, S.J. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: Clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell 1985, 41, 899–906.
- Rosenwald, A.; Wright, G.; Chan, W.C.; Connors, J.M.; Campo, E.; Fisher, R.I.; Gascoyne, R.D.; Muller-Hermelink, H.K.; Smeland, E.B.; Giltnane, J.M.; et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002, 346, 1937–1947.
- Schuetz, J.M.; Johnson, N.A.; Morin, R.D.; Scott, D.W.; Tan, K.; Ben-Nierah, S.; Boyle, M.; Slack, G.W.; Marra, M.A.; Connors, J.M.; et al. BCL2 mutations in diffuse large B-cell lymphoma. Leukemia 2012, 26, 1383–1390.
- Stilgenbauer, S.; Nickolenko, J.; Wilhelm, J.; Wolf, S.; Weitz, S.; Dohner, K.; Boehm, T.; Dohner, H.; Lichter, P. Expressed sequences as candidates for a novel tumor suppressor gene at band 13q14 in B-cell chronic lymphocytic leukemia and mantle cell lymphoma. Oncogene 1998, 16, 1891–1897.
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949.
- Robertson, L.E.; Plunkett, W.; McConnell, K.; Keating, M.J.; McDonnell, T.J. Bcl-2 expression in chronic lymphocytic leukemia and its correlation with the induction of apoptosis and clinical outcome. Leukemia 1996, 10, 456–459.
- Dohner, H.; Stilgenbauer, S.; Benner, A.; Leupolt, E.; Krober, A.; Bullinger, L.; Dohner, K.; Bentz, M.; Lichter, P. Genomic aberrations and survival in chronic lymphocytic leukemia. N. Engl. J. Med. 2000, 343, 1910–1916.
- Adams, C.M.; Mitra, R.; Gong, J.Z.; Eischen, C.M. Non-Hodgkin and Hodgkin Lymphomas Select for Overexpression of BCLW. Clin. Cancer Res. 2017, 23, 7119–7129.
- Zhou, P.; Levy, N.B.; Xie, H.; Qian, L.; Lee, C.Y.; Gascoyne, R.D.; Craig, R.W. MCL1 transgenic mice exhibit a high incidence of B-cell lymphoma manifested as a spectrum of histologic subtypes. Blood 2001, 97, 3902–3909.
- Cho-Vega, J.H.; Rassidakis, G.Z.; Admirand, J.H.; Oyarzo, M.; Ramalingam, P.; Paraguya, A.; McDonnell, T.J.; Amin, H.M.; Medeiros, L.J. MCL-1 expression in B-cell non-Hodgkin’s lymphomas. Hum. Pathol. 2004, 35, 1095–1100.
- Agarwal, B.; Naresh, K.N. Bcl-2 family of proteins in indolent B-cell non-Hodgkin’s lymphoma: Study of 116 cases. Am. J. Hematol. 2002, 70, 278–282.
- Wenzel, S.S.; Grau, M.; Mavis, C.; Hailfinger, S.; Wolf, A.; Madle, H.; Deeb, G.; Dorken, B.; Thome, M.; Lenz, P.; et al. MCL1 is deregulated in subgroups of diffuse large B-cell lymphoma. Leukemia 2013, 27, 1381–1390.
- Katz, S.G.; Labelle, J.L.; Meng, H.; Valeriano, R.P.; Fisher, J.K.; Sun, H.; Rodig, S.J.; Kleinstein, S.H.; Walensky, L.D. Mantle cell lymphoma in cyclin D1 transgenic mice with Bim-deficient B cells. Blood 2014, 123, 884–893.
- Prukova, D.; Andera, L.; Nahacka, Z.; Karolova, J.; Svaton, M.; Klanova, M.; Havranek, O.; Soukup, J.; Svobodova, K.; Zemanova, Z.; et al. Cotargeting of BCL2 with Venetoclax and MCL1 with S63845 Is Synthetically Lethal In Vivo in Relapsed Mantle Cell Lymphoma. Clin. Cancer Res. 2019, 25, 4455–4465.
- Wang, J.D.; Katz, S.G.; Morgan, E.A.; Yang, D.T.; Pan, X.; Xu, M.L. Proapoptotic protein BIM as a novel prognostic marker in mantle cell lymphoma. Hum. Pathol. 2019, 93, 54–64.
- Pfeifer, M.; Grau, M.; Lenze, D.; Wenzel, S.S.; Wolf, A.; Wollert-Wulf, B.; Dietze, K.; Nogai, H.; Storek, B.; Madle, H.; et al. PTEN loss defines a PI3K/AKT pathway-dependent germinal center subtype of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 12420–12425.
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241.
- Davis, R.E.; Brown, K.D.; Siebenlist, U.; Staudt, L.M. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J. Exp. Med. 2001, 194, 1861–1874.
- Ngo, V.N.; Davis, R.E.; Lamy, L.; Yu, X.; Zhao, H.; Lenz, G.; Lam, L.T.; Dave, S.; Yang, L.; Powell, J.; et al. A loss-of-function RNA interference screen for molecular targets in cancer. Nature 2006, 441, 106–110.
- Lenz, G.; Davis, R.E.; Ngo, V.N.; Lam, L.; George, T.C.; Wright, G.W.; Dave, S.S.; Zhao, H.; Xu, W.; Rosenwald, A.; et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 2008, 319, 1676–1679.
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119.
- Willis, T.G.; Jadayel, D.M.; Du, M.Q.; Peng, H.; Perry, A.R.; Abdul-Rauf, M.; Price, H.; Karran, L.; Majekodunmi, O.; Wlodarska, I.; et al. Bcl10 is involved in t(1;14)(p22;q32) of MALT B cell lymphoma and mutated in multiple tumor types. Cell 1999, 96, 35–45.
- Lucas, P.C.; Yonezumi, M.; Inohara, N.; McAllister-Lucas, L.M.; Abazeed, M.E.; Chen, F.F.; Yamaoka, S.; Seto, M.; Nunez, G. Bcl10 and MALT1, independent targets of chromosomal translocation in malt lymphoma, cooperate in a novel NF-kappa B signaling pathway. J. Biol. Chem. 2001, 276, 19012–19019.
- Varfolomeev, E.; Wayson, S.M.; Dixit, V.M.; Fairbrother, W.J.; Vucic, D. The inhibitor of apoptosis protein fusion c-IAP2.MALT1 stimulates NF-kappaB activation independently of TRAF1 AND TRAF2. J. Biol. Chem. 2006, 281, 29022–29029.
- Luo, J.L.; Kamata, H.; Karin, M. IKK/NF-kappaB signaling: Balancing life and death--a new approach to cancer therapy. J. Clin. Investig. 2005, 115, 2625–2632.
- Xu-Monette, Z.Y.; Medeiros, L.J.; Li, Y.; Orlowski, R.Z.; Andreeff, M.; Bueso-Ramos, C.E.; Greiner, T.C.; McDonnell, T.J.; Young, K.H. Dysfunction of the TP53 tumor suppressor gene in lymphoid malignancies. Blood 2012, 119, 3668–3683.
- Xu-Monette, Z.Y.; Wu, L.; Visco, C.; Tai, Y.C.; Tzankov, A.; Liu, W.M.; Montes-Moreno, S.; Dybkaer, K.; Chiu, A.; Orazi, A.; et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: Report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood 2012, 120, 3986–3996.
- Bea, S.; Valdes-Mas, R.; Navarro, A.; Salaverria, I.; Martin-Garcia, D.; Jares, P.; Gine, E.; Pinyol, M.; Royo, C.; Nadeu, F.; et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 18250–18255.
- Zenz, T.; Eichhorst, B.; Busch, R.; Denzel, T.; Habe, S.; Winkler, D.; Buhler, A.; Edelmann, J.; Bergmann, M.; Hopfinger, G.; et al. TP53 mutation and survival in chronic lymphocytic leukemia. J. Clin. Oncol. 2010, 28, 4473–4479.
- Sander, C.A.; Yano, T.; Clark, H.M.; Harris, C.; Longo, D.L.; Jaffe, E.S.; Raffeld, M. p53 mutation is associated with progression in follicular lymphomas. Blood 1993, 82, 1994–2004.
- Lo Coco, F.; Gaidano, G.; Louie, D.C.; Offit, K.; Chaganti, R.S.; Dalla-Favera, R. p53 mutations are associated with histologic transformation of follicular lymphoma. Blood 1993, 82, 2289–2295.
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694.
- Villunger, A.; Michalak, E.M.; Coultas, L.; Mullauer, F.; Bock, G.; Ausserlechner, M.J.; Adams, J.M.; Strasser, A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 2003, 302, 1036–1038.
- Klasa, R.J.; Gillum, A.M.; Klem, R.E.; Frankel, S.R. Oblimersen Bcl-2 antisense: Facilitating apoptosis in anticancer treatment. Antisense Nucleic Acid Drug Dev. 2002, 12, 193–213.
- O’Brien, S.; Moore, J.O.; Boyd, T.E.; Larratt, L.M.; Skotnicki, A.B.; Koziner, B.; Chanan-Khan, A.A.; Seymour, J.F.; Gribben, J.; Itri, L.M.; et al. 5-year survival in patients with relapsed or refractory chronic lymphocytic leukemia in a randomized, phase III trial of fludarabine plus cyclophosphamide with or without oblimersen. J. Clin. Oncol. 2009, 27, 5208–5212.
- Nguyen, M.; Marcellus, R.C.; Roulston, A.; Watson, M.; Serfass, L.; Murthy Madiraju, S.R.; Goulet, D.; Viallet, J.; Belec, L.; Billot, X.; et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc. Natl. Acad. Sci. USA 2007, 104, 19512–19517.
- Konopleva, M.; Watt, J.; Contractor, R.; Tsao, T.; Harris, D.; Estrov, Z.; Bornmann, W.; Kantarjian, H.; Viallet, J.; Samudio, I.; et al. Mechanisms of antileukemic activity of the novel Bcl-2 homology domain-3 mimetic GX15-070 (obatoclax). Cancer Res. 2008, 68, 3413–3420.
- Goy, A.; Berger, M.; Ford, P.; Feldman, T.; Mato, A.; Bejot, C.; Fung, H.C. Sequential single-agent obatoclax mesylate (GX15-070MS) followed by combination with rituximab in patients with previously untreated follicular lymphoma. Leuk Lymphoma 2014, 55, 2932–2934.
- Goy, A.; Hernandez-Ilzaliturri, F.J.; Kahl, B.; Ford, P.; Protomastro, E.; Berger, M. A phase I/II study of the pan Bcl-2 inhibitor obatoclax mesylate plus bortezomib for relapsed or refractory mantle cell lymphoma. Leuk Lymphoma 2014, 55, 2761–2768.
- O’Brien, S.M.; Claxton, D.F.; Crump, M.; Faderl, S.; Kipps, T.; Keating, M.J.; Viallet, J.; Cheson, B.D. Phase I study of obatoclax mesylate (GX15-070), a small molecule pan-Bcl-2 family antagonist, in patients with advanced chronic lymphocytic leukemia. Blood 2009, 113, 299–305.
- Brown, J.R.; Tesar, B.; Yu, L.; Werner, L.; Takebe, N.; Mikler, E.; Reynolds, H.M.; Thompson, C.; Fisher, D.C.; Neuberg, D.; et al. Obatoclax in combination with fludarabine and rituximab is well-tolerated and shows promising clinical activity in relapsed chronic lymphocytic leukemia. Leuk Lymphoma 2015, 56, 3336–3342.
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681.
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428.
- Wilson, W.H.; O’Connor, O.A.; Czuczman, M.S.; LaCasce, A.S.; Gerecitano, J.F.; Leonard, J.P.; Tulpule, A.; Dunleavy, K.; Xiong, H.; Chiu, Y.L.; et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: A phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010, 11, 1149–1159.
- Roberts, A.W.; Seymour, J.F.; Brown, J.R.; Wierda, W.G.; Kipps, T.J.; Khaw, S.L.; Carney, D.A.; He, S.Z.; Huang, D.C.; Xiong, H.; et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: Results of a phase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. 2012, 30, 488–496.
- Vogler, M.; Hamali, H.A.; Sun, X.M.; Bampton, E.T.; Dinsdale, D.; Snowden, R.T.; Dyer, M.J.; Goodall, A.H.; Cohen, G.M. BCL2/BCL-X(L) inhibition induces apoptosis, disrupts cellular calcium homeostasis, and prevents platelet activation. Blood 2011, 117, 7145–7154.
- Zhang, H.; Nimmer, P.M.; Tahir, S.K.; Chen, J.; Fryer, R.M.; Hahn, K.R.; Iciek, L.A.; Morgan, S.J.; Nasarre, M.C.; Nelson, R.; et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007, 14, 943–951.
- Kipps, T.J.; Eradat, H.; Grosicki, S.; Catalano, J.; Cosolo, W.; Dyagil, I.S.; Yalamanchili, S.; Chai, A.; Sahasranaman, S.; Punnoose, E.; et al. A phase 2 study of the BH3 mimetic BCL2 inhibitor navitoclax (ABT-263) with or without rituximab, in previously untreated B-cell chronic lymphocytic leukemia. Leuk Lymphoma 2015, 56, 2826–2833.
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208.
More
Information
Subjects:
Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.0K
Revisions:
2 times
(View History)
Update Date:
23 Jun 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No