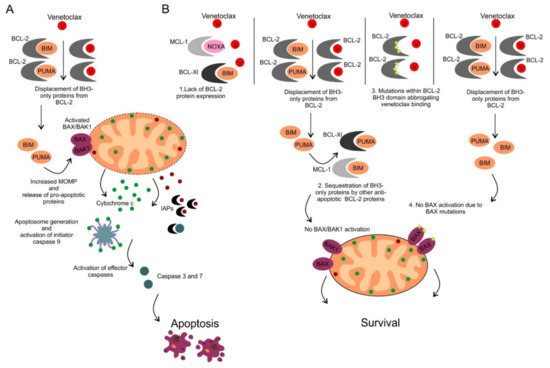
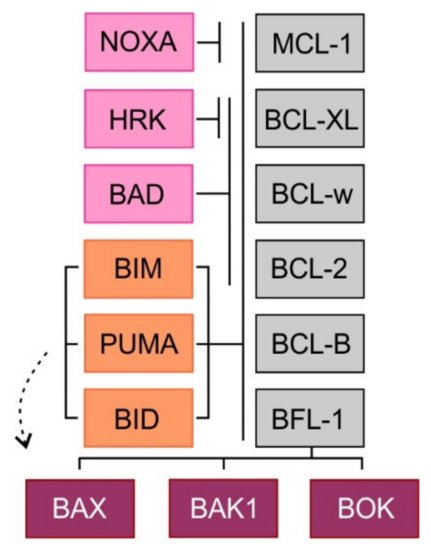
The ability to inhibit mitochondrial apoptosis is a hallmark of B-cell non-Hodgkin lymphomas (B-NHL). Activation of mitochondrial apoptosis is tightly controlled by members of B-cell leukemia/lymphoma-2 (BCL-2) family proteins via protein-protein interactions. Altering the balance between anti-apoptotic and pro-apoptotic BCL-2 proteins leads to apoptosis evasion and extended survival of malignant cells. The pro-survival BCL-2 proteins: B-cell leukemia/lymphoma-2 (BCL-2/BCL2), myeloid cell leukemia-1 (MCL-1/MCL1) and B-cell lymphoma-extra large (BCL-XL/BCL2L1) are frequently (over)expressed in B-NHL, which plays a crucial role in lymphoma pathogenesis, disease progression, and drug resistance.
- apoptosis
- non-Hodgkin lymphomas (NHL)
- B-cell leukemia/lymphoma-2 (BCL-2)
- venetoclax
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
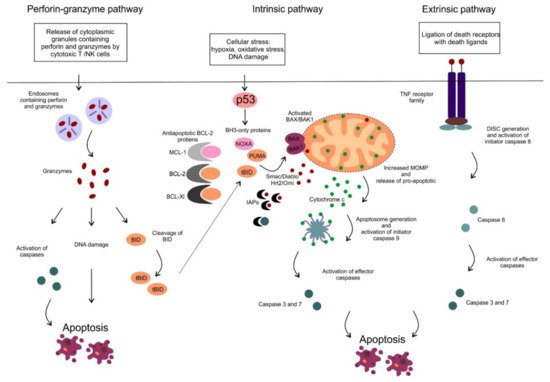
2. Mitochondrial Apoptotic Pathway
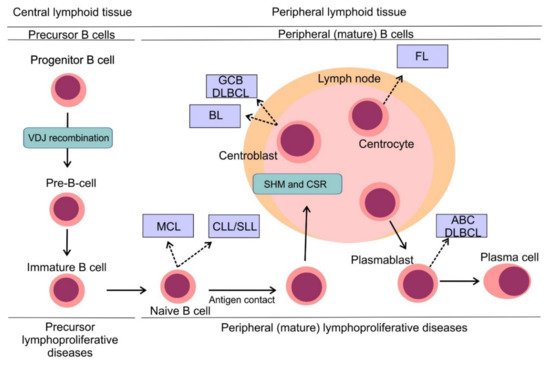
3. B-Cell Non-Hodgkin Lymphomas: Pathogenesis and Classification
4. Deregulation of BCL-2 Proteins in B-Cell Non-Hodgkin Lymphomas
| B-NHL Subtype | BCL-2 Positivity * | Mechanism of BCL-2 Overexpression |
|---|---|---|
| DLBCL | 49%–67% [30][31][32][30,31,32] | BCL2 translocation in GCB DLBCL (30% of cases) [33] BCL2 amplification in ABC DLBCL (20% of cases) [34] |
| FL | > 90% [35][36][37][35,36,37] | BCL2 translocation (90% of cases) [35][36][35,36] |
| MCL | BCL-2 positive [38] | 13q14.3 loss (55% of cases) [39] 18q21 gains (20% of cases) [37][39][37,39] |
| BL | BCL-2 negative, weak expression in up to 20% [40] | NA |
| CLL/SLL | BCL-2 positive (high expression in majority of the cases) [41] | 13q14.3 loss (68% cases) [42] BCL2 gene hypomethylation [43] BCL2 translocation (rare, < 5%) [41] |
| MZL | >80% [44] | unknown |
5. Therapeutic Inhibition of Anti-Apoptotic BCL-2 Proteins