 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | David Gallego-Ortega | + 2761 word(s) | 2761 | 2021-05-19 05:55:13 | | | |
| 2 | Vivi Li | Meta information modification | 2761 | 2021-05-25 08:07:15 | | |
Video Upload Options
The emergence of immunotherapy has been an astounding breakthrough in cancer treatments. In particular, immune checkpoint inhibitors, targeting PD-1 and CTLA-4, have shown remarkable therapeutic outcomes. However, response rates from immunotherapy have been reported to be varied, with some having pronounced success and others with minimal to no clinical benefit. An important aspect associated with this discrepancy in patient response is the immune-suppressive effects elicited by the tumour microenvironment (TME). Immune suppression plays a pivotal role in regulating cancer progression, metastasis, and reducing immunotherapy success. Most notably, myeloid-derived suppressor cells (MDSC), a heterogeneous population of immature myeloid cells, have potent mechanisms to inhibit T-cell and NK-cell activity to promote tumour growth, development of the pre-metastatic niche, and contribute to resistance to immunotherapy. Accumulating research indicates that MDSC can be a therapeutic target to alleviate their pro-tumourigenic functions and immunosuppressive activities to bolster the efficacy of checkpoint inhibitors.
1. Introduction
2. Targeting MDSCs in Cancer
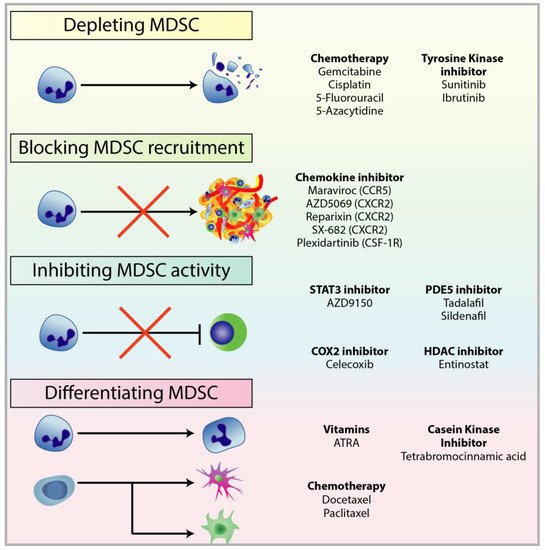
2.1. Depleting MDSC Populations
2.2. Blockade of MDSC Migration
2.3. Attenuating MDSC Immunosuppressive Functions
2.4. Inducing MDSC Differentiation
3. Combining MDSC-Targeted Treatments with Immunotherapy
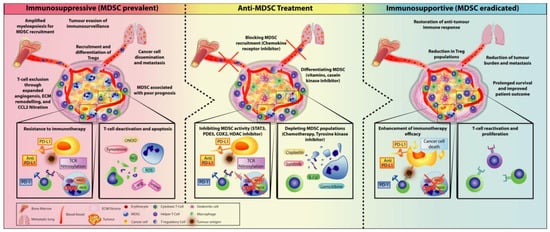
3.1. Checkpoint Inhibitors Combined with MDSC Depletion
3.2. Immunotherapy Combined with Obstructing MDSC Trafficking Therapy
References
- Garcia-Lora, A.; Algarra, I.; Garrido, F. MHC class I antigens, immune surveillance, and tumor immune escape. J. Cell Physiol. 2003, 195, 346–355.
- Fernald, K.; Kurokawa, M. Evading apoptosis in cancer. Trends Cell Biol. 2013, 23, 620–633.
- Burkholder, B. Tumor-induced perturbations of cytokines and immune cell networks. Biochim. Biophys. Acta 2014, 1845, 182–201.
- Salgado, R. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: Recommendations by an International TILs Working Group 2014. Ann. Oncol. 2015, 26, 259–271.
- Chow, M.T.; Moller, A.; Smyth, M.J. Inflammation and immune surveillance in cancer. Semin Cancer Biol. 2012, 22, 23–32.
- Valdes-Mora, F. Single-Cell Transcriptomics in Cancer Immunobiology: The Future of Precision Oncology. Front. Immunol. 2018, 9, 2582.
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268.
- Messmer, M.N. Tumor-induced myeloid dysfunction and its implications for cancer immunotherapy. Cancer Immunol. Immunother. 2015, 64, 1–13.
- Mantovani, A. The growing diversity and spectrum of action of myeloid-derived suppressor cells. Eur. J. Immunol. 2010, 40, 3317–3320.
- Diaz-Montero, C.M. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2009, 58, 49–59.
- Highfill, S.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl Med. 2014, 6, 237ra67.
- Meyer, C. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol. Immunother. 2014, 63, 247–257.
- Tarhini, A.A. Immune monitoring of the circulation and the tumor microenvironment in patients with regionally advanced melanoma receiving neoadjuvant ipilimumab. PLoS ONE 2014, 9, e87705.
- Weber, J. Phase I/II Study of Metastatic Melanoma Patients Treated with Nivolumab Who Had Progressed after Ipilimumab. Cancer Immunol. Res. 2016, 4, 345–353.
- Groth, C. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25.
- Suzuki, E. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin. Cancer Res. 2005, 11, 6713–6721.
- Sevko, A. Antitumor effect of paclitaxel is mediated by inhibition of myeloid-derived suppressor cells and chronic inflammation in the spontaneous melanoma model. J. Immunol. 2013, 190, 2464–2471.
- Eriksson, E. Gemcitabine reduces MDSCs, tregs and TGFbeta-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J. Transl. Med. 2016, 14, 282.
- Vincent, J. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010, 70, 3052–3061.
- Mikyskova, R. Cyclophosphamide-induced myeloid-derived suppressor cell population is immunosuppressive but not identical to myeloid-derived suppressor cells induced by growing TC-1 tumors. J. Immunother. 2012, 35, 374–384.
- Iida, Y. Contrasting effects of cyclophosphamide on anti-CTL-associated protein 4 blockade therapy in two mouse tumor models. Cancer Sci. 2017, 108, 1974–1984.
- Fleming, V. Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front. Immunol. 2018, 9, 398.
- Ko, J.S. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin. Cancer Res. 2009, 15, 2148–2157.
- Kodera, Y. Sunitinib inhibits lymphatic endothelial cell functions and lymph node metastasis in a breast cancer model through inhibition of vascular endothelial growth factor receptor 3. Breast Cancer Res. 2011, 13, R66.
- Qin, H. Generation of a new therapeutic peptide that depletes myeloid-derived suppressor cells in tumor-bearing mice. Nat. Med. 2014, 20, 676–681.
- Weber, R. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 1310.
- Blattner, C. CCR5(+) Myeloid-Derived Suppressor Cells Are Enriched and Activated in Melanoma Lesions. Cancer Res. 2018, 78, 157–167.
- Robinson, S.C. A chemokine receptor antagonist inhibits experimental breast tumor growth. Cancer Res. 2003, 63, 8360–8365.
- Velasco-Velazquez, M. CCR5 antagonist blocks metastasis of basal breast cancer cells. Cancer Res. 2012, 72, 3839–3850.
- Katoh, H. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell 2013, 24, 631–644.
- Toh, B. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 2011, 9, e1001162.
- Zhu, H. CXCR2(+) MDSCs promote breast cancer progression by inducing EMT and activated T cell exhaustion. Oncotarget 2017, 8, 114554–114567.
- Steele, C.W. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845.
- Di Mitri, D. Tumour-infiltrating Gr-1+ myeloid cells antagonize senescence in cancer. Nature 2014, 515, 134–137.
- Sun, L. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight 2019, 4.
- Holmgaard, R.B. Targeting myeloid-derived suppressor cells with colony stimulating factor-1 receptor blockade can reverse immune resistance to immunotherapy in indoleamine 2,3-dioxygenase-expressing tumors. EBioMedicine 2016, 6, 50–58.
- Zhu, Y. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069.
- Richardsen, E. Macrophage-colony stimulating factor (CSF1) predicts breast cancer progression and mortality. AntiCancer Res. 2015, 35, 865–874.
- Sluijter, M. Inhibition of CSF-1R supports T-cell mediated melanoma therapy. PLoS ONE 2014, 9, e104230.
- Mok, S. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 2014, 74, 153–161.
- Kumar, V. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell 2017, 32, 654–668.
- Elliott, L.A. Human Tumor-Infiltrating Myeloid Cells: Phenotypic and Functional Diversity. Front. Immunol. 2017, 8, 86.
- Sinha, P. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007, 67, 4507–4513.
- Eruslanov, E. Pivotal Advance: Tumor-mediated induction of myeloid-derived suppressor cells and M2-polarized macrophages by altering intracellular PGE(2) catabolism in myeloid cells. J. Leukoc. Biol. 2010, 88, 839–848.
- Ochoa, A.C. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin. Cancer Res. 2007, 13, 721s–726s.
- Veltman, J.D. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. EBioMedicine 2010, 10, 464.
- Obermajer, N. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood 2011, 118, 5498–5505.
- Zelenay, S. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270.
- Serafini, P. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J. Exp. Med. 2006, 203, 2691–2702.
- Lin, S. Phosphodiesterase-5 inhibition suppresses colonic inflammation-induced tumorigenesis via blocking the recruitment of MDSC. Am. J. Cancer Res. 2017, 7, 41–52.
- Tai, L.H. Phosphodiesterase-5 inhibition reduces postoperative metastatic disease by targeting surgery-induced myeloid derived suppressor cell-dependent inhibition of Natural Killer cell cytotoxicity. Oncoimmunology 2018, 7, e1431082.
- Califano, J.A. Tadalafil augments tumor specific immunity in patients with head and neck squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 30–38.
- Hassel, J.C. Tadalafil has biologic activity in human melanoma. Results of a pilot trial with Tadalafil in patients with metastatic Melanoma (TaMe). Oncoimmunology 2017, 6, e1326440.
- Weed, D.T. Tadalafil reduces myeloid-derived suppressor cells and regulatory T cells and promotes tumor immunity in patients with head and neck squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 39–48.
- Ohl, K.; Tenbrock, K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front. Immunol. 2018, 9, 2499.
- Wang, Y.Y. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: An update on its pharmacokinetic and pharmacodynamic properties. Drug Des. Devel. Ther. 2014, 8, 2075–2088.
- Hiramoto, K. Myeloid lineage-specific deletion of antioxidant system enhances tumor metastasis. Cancer Prev. Res. (Phila) 2014, 7, 835–844.
- Nagaraj, S. Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin. Cancer Res. 2010, 16, 1812–1823.
- Molon, B. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J. Exp. Med. 2011, 208, 1949–1962.
- De Santo, C. Nitroaspirin corrects immune dysfunction in tumor-bearing hosts and promotes tumor eradication by cancer vaccination. Proc. Natl. Acad. Sci. USA 2005, 102, 4185–4190.
- Reilley, M.J. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: Results of a phase 1b trial. J. Immunother. Cancer 2018, 6, 119.
- Kusmartsev, S. Reversal of myeloid cell-mediated immunosuppression in patients with metastatic renal cell carcinoma. Clin. Cancer Res. 2008, 14, 8270–8278.
- Mirza, N. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006, 66, 9299–9307.
- Iclozan, C. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol. Immunother. 2013, 62, 909–918.
- Nefedova, Y. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. 2007, 67, 11021–11028.
- Cheng, P. Effects of notch signaling on regulation of myeloid cell differentiation in cancer. Cancer Res. 2014, 74, 141–152.
- Orillion, A. Entinostat Neutralizes Myeloid-Derived Suppressor Cells and Enhances the Antitumor Effect of PD-1 Inhibition in Murine Models of Lung and Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 5187–5201.
- Christmas, B.J. Entinostat Converts Immune-Resistant Breast and Pancreatic Cancers into Checkpoint-Responsive Tumors by Reprogramming Tumor-Infiltrating MDSCs. Cancer Immunol. Res. 2018, 6, 1561–1577.
- Mikyskova, R. DNA demethylating agent 5-azacytidine inhibits myeloid-derived suppressor cells induced by tumor growth and cyclophosphamide treatment. J. Leukoc. Biol. 2014, 95, 743–753.
- Kodumudi, K.N. A novel chemoimmunomodulating property of docetaxel: Suppression of myeloid-derived suppressor cells in tumor bearers. Clin. Cancer Res. 2010, 16, 4583–4594.
- Michels, T. Paclitaxel promotes differentiation of myeloid-derived suppressor cells into dendritic cells in vitro in a TLR4-independent manner. J. Immunotoxicol. 2012, 9, 292–300.
- Law, A.M. The innate and adaptive infiltrating immune systems as targets for breast cancer immunotherapy. Endocr. Relat. Cancer 2017, 24, R123–R144.
- Kim, K. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11774–11779.


