 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Cátia Teixeira | + 3715 word(s) | 3715 | 2021-02-15 08:31:56 | | | |
| 2 | Catherine Yang | Meta information modification | 3715 | 2021-03-19 01:57:53 | | |
Video Upload Options
Malaria is among the deadliest infectious diseases in the world caused by Plasmodium parasites.
1. Introduction
A century has elapsed since Laveran described Plasmodium parasites and Ross confirmed that they were transmitted by mosquitoes [1]. Still, malaria remains a leading cause of mortality and morbidity worldwide [2]. Although we have been witnessing the decrease in malaria burden over the last decade, prospects for malaria eradication are now threatened by resistance to artemisinin-based combination therapies, the current first-line antimalarial treatment [3], urging the development of new classes of antimalarials. While a vaccine remains elusive, we depend on chemotherapeutic agents to both treat infections and prevent disease. Most available antiplasmodials target the pathogenic blood stages in humans [4]. However, to eradicate malaria, it is mandatory to develop compounds that block parasite transmission, cure the asymptomatic hepatic infection and clear the latent forms in the liver [5]. Therefore, elimination efforts require identification of new drug classes acting at multiple stages of the parasite life cycle. One strategy to accelerate development of antimalarials is to recycle known drug scaffolds [4].
Acridine (AC, Figure 1) derivatives have attracted much attention due to their broad spectrum of biological activity, such as antimicrobial, antitumoral, anti-Alzheimer, antiprionic, antileishmanial and antimalarials agents [4][6][7]. Although different modes of action seem to be exerted by AC derivatives depending on their therapeutic targets, the most consensual mechanism of action (MOA) of AC analogues against different diseases is interaction with DNA [8]. However, other MOAs have been suggested to elucidate the antimalarial activity of AC derivatives, such as inhibition of the parasite’s (i) type II topoisomerases, (ii) mitochondrial bc1 complex, or (iii) hemozoin formation [4]. The interest in AC-based structures as antimalarials occurred from early findings on antiprotozoan/antimicrobial activity of the classic synthetic dyes methylene blue (MB, Figure 1) and, later, acridine orange (ACO, Figure 1) with IC50 values of 7.8 and 465.7 nM (for MB and ACO, respectively) against Plasmodium falciparum (Pf) 3D7 strain [9]. Both the tendency to induce skin coloration and toxicity of MB soon triggered the pursuit for more suitable antimalarial substitutes, which led to the synthesis of quinacrine (QN, Figure 1), in 1932 [10]. QN was thus the first clinically tested synthetic antimalarial drug and was widely used as an antimalarial by soldiers during WWII but was soon superseded by chloroquine (CQ, Figure 1), whose bioavailability, safety and efficiency were substantially superior [4][11]. However, with the widespread resistance of Pf to CQ, the search for more efficient QN derivatives or analogues remains vigorous. Amongst different approaches reported in the literature, chemical modifications and the combination or conjugation of AC-based compounds with known relevant pharmacophores have been addressed, in an attempt to outwit resistance mechanisms to classical antimalarials and to produce safer and synergistic or multi-target action compounds. In this context, the present review will focus on efforts made in the last two decades for the development of AC-based compounds as a tool for the rescuing or repurposing of the classical antimalarial quinacrine.
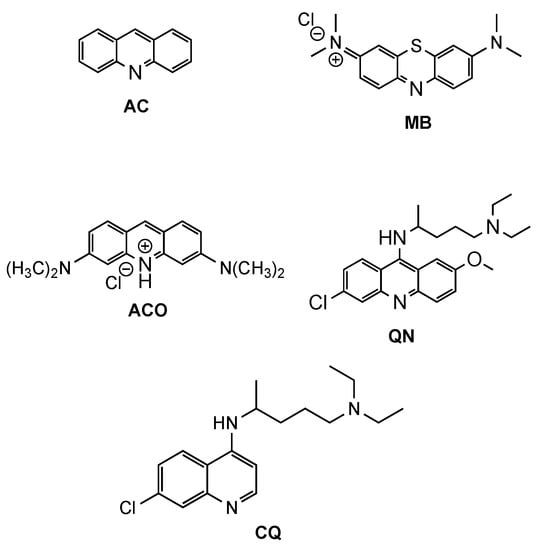
Figure 1. Chemical structure of acridine (AC), methylene blue (MB), acridine orange (ACO), quinacrine (QN) and chloroquine (CQ).
2. Hybrids Containing the 9-Aminoacridine Scaffold
Another noteworthy approach used for recycling the QN scaffold and obtaining faster and available new antimalarial treatments is based on the concept of hybrid drugs. A hybrid molecule is formed through the covalent link between two known chemical moieties with different biological modes of action. This methodology was developed aiming to obtain molecules with better biologic activity, solubility profile and stability than the parent drugs, as well as less susceptibility to the development of drug resistance (Scheme 1). In the last decade, this strategy has attracted attention, mainly in the field of the development of antimalarial drugs due to the urgency of novel compounds to fight malaria disease [12][13].
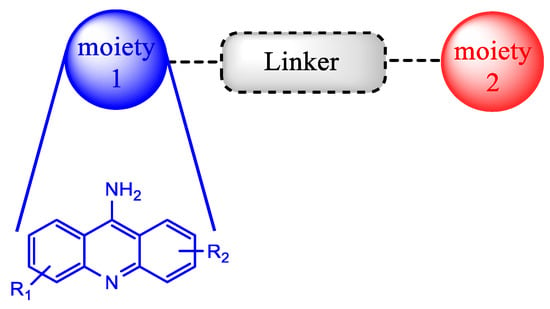
Scheme 1. Representative scheme of a QN hybrid drug.
2.1. Bisacridine Hybrids
Bisacridines were among the first classes of hybrids extensively explored as DNA intercalators. These hybrids are composed of two acridine portions, and studies suggested that the length and flexibility of the spacer between these two acridines affect the intercalation property. The first findings related to this class of compounds came from the work of King et al. [14]. Using spectrophotometric and viscosimetric experiments, they found that a longer and more flexible spacer favored bis-intercalation, while bisacridines with a shorter and less flexible spacer monointercalated [14]. The potential of bisacridines as antimalarials was first reported by Girault et al. [15], who developed three series of bisacridines by linking two acridine cores through three different spacers: alkanediamines, linear polyamines or branched polyamines. The in vitro antimalarial activity of all the compounds against a CQR Pf FcB1R strain, as well their cytotoxic effects on MRC-5 cells, were evaluated. The best ones (Figure 2) were further evaluated for their efficiency to inhibit the growth of another seven Pf strains (3D7, F32a, GP1, FCR3, FCM29, W2 and K1). Compound 17a (Figure 2), a piperazine derivative, showed the best results with high and selective activity against different Pf strains (IC50 values ranging from 8 to 18 nM), and a total absence of cytotoxic effects upon MRC-5 cells and murine macrophages [15].
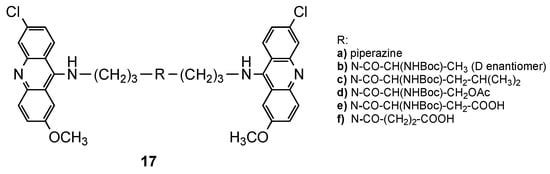
Figure 2. The best compounds 17 developed by Girault and co-workers [15].
From their previous work [15], they selected a set of compounds based on the cytotoxicity/activity ratio (17a–f) and tested their activity upon mice infected by P. berghei at the concentration of 40 mg/kg [16]. Two additional compounds (18a and 18b, Figure 3) were synthetized to verify the influence of the bond between the side chain and the polyamine linker on antimalarial activity and cytotoxicity. The lack of solubility of compound 17a prevent it from being assessed in vivo; however, important considerations were obtained for the other compounds: (i) the most active compounds in vitro [15] (17a–f) were found to be inactive at the tested concentration; (ii) the N-alkylation of the central amino group (18b), instead of N-acylated (18a), generated toxicity in vivo; (iii) the compound 18a was the most active compound in vivo, with 22% of prolongation of the mean survival time. These results indicated the difficulty to predict relationships between in vitro and in vivo activity and toxicity, even between members of the same family of compounds [16].
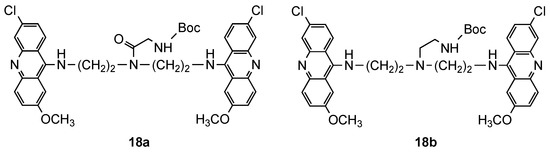
Figure 3. Compounds 18 developed by Girault and co-workers [16].
Another library of bisacridines was developed by Caffrey et al. [17]. The authors synthesized 16 bisacridines holding different linkers (polyamine, alkylated polyamine, acylated polyamine, alkyl or polyether) and two bis-aza-acridines with a polyamine and alkyl linker. The compounds were evaluated for their in vitro antimalarial activity against the CQS Pf 3D7 and the CQR Pf W2 strains, and their cytotoxicity on HL-60 mammalian cell lines. Structure–activity studies demonstrated some interesting aspects: (i) an increase higher than the minimum linker length (10 Å in this case) increases the activity against the parasite, but also increases the cytotoxicity; on the other hand, (ii) the addition of steric bulk and conformational constraint in the polyamine linker through alkylation, acylation or substitution of polyamine-reduced cytotoxicity retains antimalarial activity; (iii) the polyether linker improves potency relative to the equivalent polyamine linker as well as the substitution of the acridine heterocycle by the aza-acridine.
Although most of the compounds presented EC50 values comparable to references CQ and QN [17], compound 19 (Figure 4), a piperazinyl derivative, displayed the best therapeutic indices (3D7: EC50 = 64 nM; W2: EC50 = 112 nM), which supported the results of Girault and colleagues [15][16].
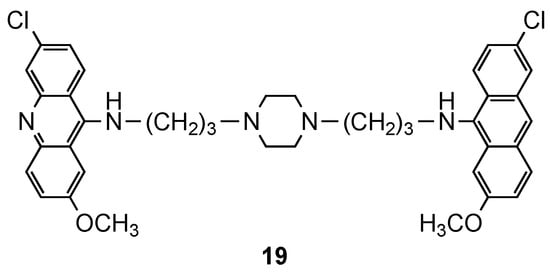
Figure 4. Compound 19 developed by Caffrey and co-workers [17].
2.2. Acridine–Artemisinin Hybrids
The relevance of artemisinin-based antimalarials in the current clinical strategies against CQR malaria, and the recent emergence of resistance to ACT [3], also led to the exploration of several hybrid molecules containing artemisinin moiety. The exact mechanism of this first line antimalarial and its derivatives is not completely understood, but it is believed to rely on the cleavage of the endoperoxide bond by reduced Fe2+, leading to cytotoxic carbon-centered free radicals and the alkylation of critical biomolecules of the parasite [18][19][20]. In addition to the intrinsic antimalarial activity of the acridine derivatives, their conjugation with artemisinin scaffold is expected to increase the accumulation of the hybrid in the food vacuole, and consequently, the efficiency of artemisinin [21]. One of the first works reporting such hybrids was carried out by Jones et al. [22]. In this study, 1,2,4-trioxane, derived from artemisinin, was covalently linked to 9-diaminoalkyl-6-chloro-2-methoxyacridine moieties through two different spacers (20, and 21, Figure 5). The compounds were evaluated against a CQS Pf strain (3D7) and presented IC50 values ranging from 5.96 to 289.52 nM. However, even the best compound (20a, Figure 5) was less potent than the controls (IC50(20a) = 5.96 nM; IC50(dihydroartemisinin) = 2.30 nM; IC50(artemether) = 3.53 nM) [22].
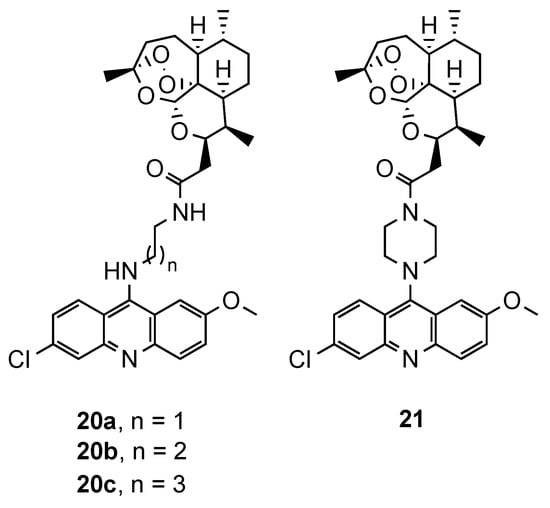
Figure 5. Compounds (20 and 21) developed by Jones and co-workers [22].
In a similar study, Araújo et al. [21] described the synthesis of a novel series of semi-synthetic trioxaquines and synthetic trioxolaquines, both covalently linked to the 9-diaminoalkyl-6-chloro-2-methoxyacridine moiety (22 and 23, Figure 6). The in vitro antimalarial activity, against CQS and CQR Pf strains, was evaluated, and both series of compounds were active in a low nanomolar range. In general, the synthetic series were more potent than the semi-synthetic artemisinin derivatives. Compound 22a (Figure 6) displayed the best results in the semi-synthetic series (IC50(3D7) = 12.52 nM; IC50(K1) = 14.34 nM), while compound 23a (Figure 6) was the best in synthetic series (IC50(3D7) = 9.67 nM; IC50(K1) = 7.20 nM). Both compounds have -(CH2)2− as a linker, which shows that the activity is very dependent on the nature and length of the linker [21].
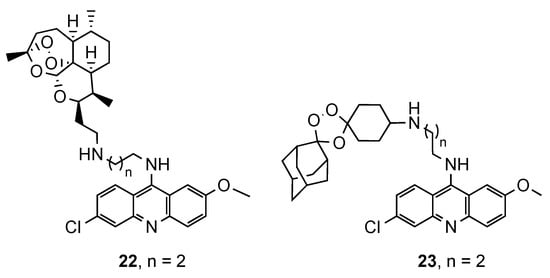
Figure 6. Synthetic and semi-synthetic artemisinin derivatives 22 and 23 developed by Araújo and co-workers [21].
In the same line of thought, Joubert et al. reported the synthesis and in vitro antimalarial activity of hybrids obtained by covalently linking artemisinin and acridine pharmacophores through an aminoethyl ether linker [23]. Most of the compounds were shown to be more active than the reference CQ, but less than the references dihydroartemisinin (DHA) and artesunate against CQS and CQR Pf strains. Hybrid 24 (Figure 7), which contains ehtylenediamine as a linker, was the most promising antimalarial compound with 7-fold higher antimalarial potency than CQ against both strains, along with a high selectivity index towards the parasitic cells (24: IC50(NF54) = 2.6 nM; IC50(Dd2) = 35.3 nM; CQ: IC50(NF54) = 18.5 nM; IC50(Dd2) = 271.7 nM; SI = 615). It is noteworthy that the hybrids possessed generally higher activity than their precursor 9-aminoacridines and less cytotoxic for human cells than DHA [23].
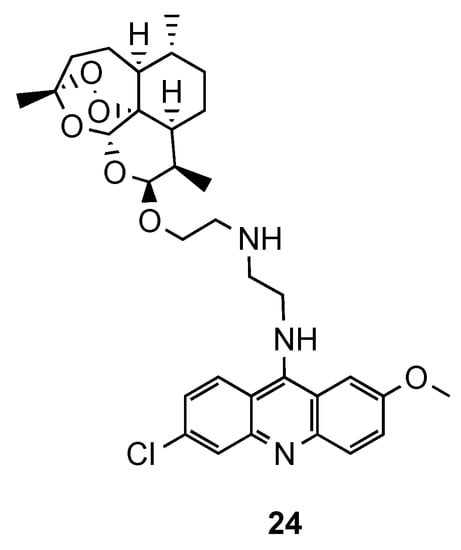
Figure 7. Most active compound 24 developed by Joubert and co-workers [23].
2.3. Acridine–Aminoquinoline Hybrids
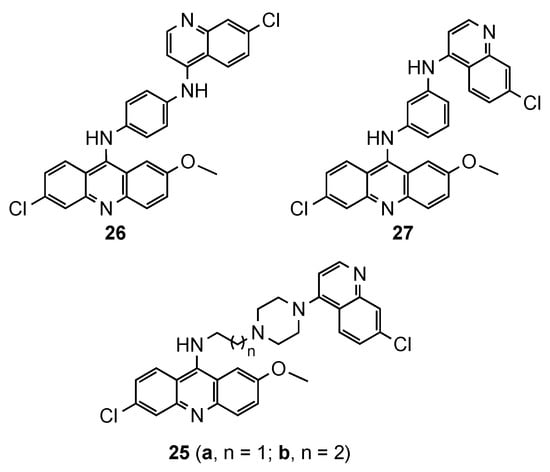
Despite the emergence of resistance to 4-aminoquinoline drugs, this pharmacophore has a crucial role inhibiting the heme polymerization and parasite growing. It is believed that the steric hindrance in bulky compounds, such as bisquinolines, would avoid the drug efflux and overcome the resistance [24][25][26]. However, hybrids containing the acridine and quinoline moieties are scarcely reported. This prominent approach was described by Kumar et al. [27], who synthesized quinoline–acridine hybrids covalently linked by (i) an alkyldiamine chain functionalized piperazine (25a and 25b, Figure 8), or (ii) a p/m-phenylenediamine (26 and 27, Figure 8). Compound 27 exhibited the best in vitro antimalarial activity against the Pf NF54 strain (MIC = 0.25 μg/mL), but still less potent than the reference CQ (MIC = 0.125 μg/mL). This compound was tested in vivo in Swiss mice infected with the CQR N-67 strain of Plasmodium yoelii, and the results displayed the complete clearance of parasitemia on day 4 at the dose of 50 mg/kg × 4 days by intraperitonial route. However, none of mice survived beyond day 28 [27].
2.4. Acridine–Chalcone Hybrids
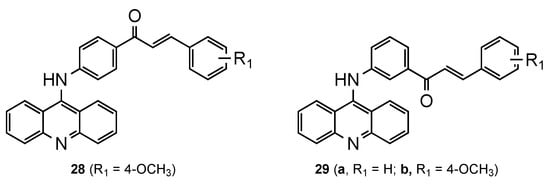
Based on reports of the diverse range of pharmacological activities of chalcones, namely, their ability to defeat plasmodium parasites [28][29], Tomar et al. [30] developed a series of new 9-acridinylamino chalcone derivatives (28–29, Figure 9). The analogues presented different substituents (-NO2, -NH2, -OH, -CH3, -OCH3, -Cl) on chalcone’s ring B at different positions. To complement the structure–activity relationship study, chalcone’s ring A was linked to the 9-aminoacridine in positions 3 or 4. All the compounds were screened for in vitro antimalarial activity against the CQS Pf NF54 strain, and it was evident that the location and nature of the substituent(s) on chalcone’s ring B derivatives are essential. The eleven compounds fully inhibited the maturation of parasite at a concentration greater than or equal to 10 μg/mL, namely, compounds 28 and 29a–b exhibited a percentage inhibition above 70% at 2 μg/mL concentration. These three acridine–chalcone hybrids were further screened for in vivo efficacy against a CQR rodent malaria parasite Plasmodium yoelii (strain N-67) in a Swiss mice model. However, no significant inhibition in parasitemia was observed [30].
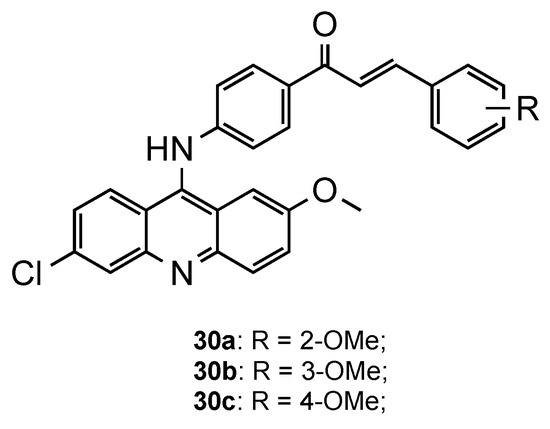
Following the same trend, Prajapati et al. developed another series of acridine–chalcone hybrids [31]. In this case, the authors included the substituents chlorine and methoxy, known to increase antimalarial activity, at positions 6 and 2 of the acridine ring, respectively. Additionally, they also explored the influence of the substituents and respective position on chalcone’s ring B. The in vitro antimalarial activity was next assessed against CQS Pf 3D7 and CQR Pf Dd2 strains, and the cytotoxicity against the HeLa cell line. Overall, structure–activity results suggested that the monomethoxy substitution significantly increased antimalarial activity, mainly at the 4-position of the ring, when compared to other tested substituents, which supports the results of Tomar and colleagues [30]. Compounds 30a–c (Figure 10) were the most potent, with IC50 values in the range of 0.30–0.52 μM and 0.15–0.32 μM against the Pf 3D7 and Pf Dd2 strains, respectively. Additionally, all the compounds exhibited high selective and resistance indices [31].
2.5. Acridine–Cinnamoyl Hybrids
The potential of classical antimalarial drugs conjugated with cinnamoyl moiety was exhaustively explored by Pérez et al. [32][33][34][35][36]. In one of the studies, the authors reported a new class of hybrids combining the acridine core with a cinnamoyl moiety through an aminobutyl chain [35]. The compounds displayed mid-nanomolar in vitro activity against erythrocytic stages of the CQR Pf W2 strain (126 nM < IC50 < 345 nM). Compound 31a (Figure 11) showed not only a similar activity in the blood stage (IC50 = 138 nM) to CQ (IC50 = 138 nM), as it also presented activity against hepatic forms of P. berghei three times higher (IC50 = 3.2 μM) than the reference PQ (IC50 = 8 μM). At the time, this was an unprecedented result, since QN derivatives were described as devoid of activity against Plasmodium hepatic forms. Moreover, all the compounds proved to be non-toxic when tested in vitro on Huh7 human hepatoma cells [35].
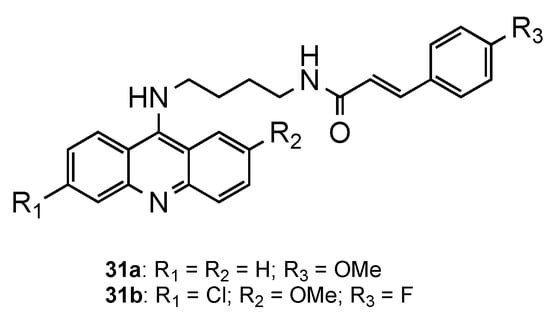
Later, the same authors [36] synthesized the same hybrids mentioned above, but including the 6-chloro and 2-methoxy substituents on the aminoacridine core (Figure 12). The results of the in vitro antimalarial activity confirmed once more that these substituents significantly improved the antimalarial activity against blood-stage parasites with promising antimalarial activities against CQS Pf 3D7 and CQR Pf Dd2 strains. The best compound (31b, Figure 12) exhibited (i) comparable or better activities than CQ against the blood forms of the Pf strains 3D7, Dd2 and W2 (31b: IC50(3D7) = 29.8 nM, IC50(Dd2) = 131.0 nM, IC50(W2) = 17.8 nM; CQ: IC50(3D7) = 21.0 nM, IC50(Dd2) = 107.5 nM, IC50(W2) = 225.8 nM); ii) better activity against the P. berghei hepatic stage than PQ (31b: IC50 = 1.6 μM; PQ: IC50 = 7.5 μM); and iii) low cytotoxicity against human HepG2 cells (SI = 1257) [36].
2.6. Other Acridine Hybrids
The synthesis of hybrids containing the acridine and clotrimazole-like pharmacophore was first described by Sandra Gemma et al. [37][38]. In previous works [37][38], the authors changed the chemical structure of clotrimazole, an antimycotic drug, to improve its antimalarial activity. In their more recent work [39], this resulting compound was next linked to 9-aminoacridine and assessed against three Pf CQS (D10, 3D7 and NF54) and two CQR (W2 and K1) strains. In the in vitro assays, except for the CQS D10 strain, compound 32 (Figure 12) exhibited better activities (IC50 values ranging from 1.0 to 49 nM for CQS D10, 3D7, and NF54 strains and from 9.0 to 59 nM for CQR W2, and K1 strains) than the reference CQ. This compound has a polyarylmethyl, which gives it the ability to selectively interact with the free heme-iron center and consequently accumulate into the food vacuole, promoting the generation of toxic radical species for plasmodium’s parasites [39].
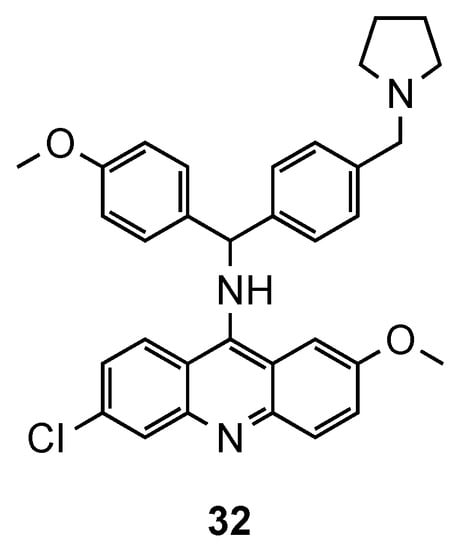
Figure 12. Compound 32 developed by Gemma and co-workers et al. [39].
Kumar et al. directed their attention to triazine–acridine hybrids (33, Figure 13) [40]. The synthesized compounds were evaluated for their in vitro antimalarial activity against the CQS Pf strain and their cytotoxicity on the VERO cell line. Of the whole set, compounds 33a–c exhibited good antimalarial activities in vitro with a high selectivity index, some of them being better than CQ (33a: IC50 = 6.97 nM, SI = 2896.02; 33b: IC50 = 4.21 nM, SI = 295.02; 33c: IC50 = 4.27 nM, SI = 315.39; CQ: IC50 = 8.15 nM, SI = 8983). Compound 33a was further subjected to in vivo study against the CQR N-67 strain of P. yoelii orally in Swiss mice at a dose of 100 mg/kg for four days. Although the compound could suppress the infection in 96.59%, it could not provide significant protection to the treated mice in 28 days survival assay [40].
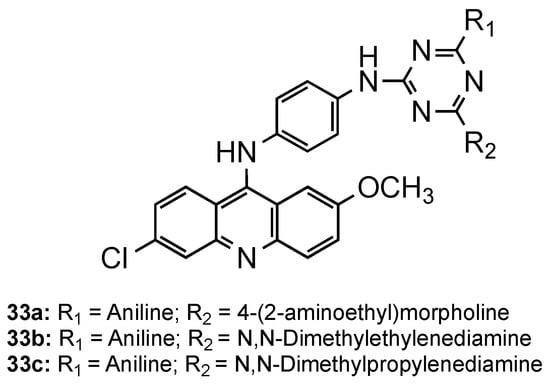
Figure 13. Triazine–acridine hybrids 33 developed by Kumar and co-workers [40].
Based on reports concerning the good inhibitory activity of steroidal 4-aminoquinolines, and adamantyl-aminoquinolines [41], Tot et al. [42] expanded their work towards the synthesis and in vitro antimalarial activity of news 9-aminoalkylaminoacridine derivatives possessing steroid and adamantane carriers (34 and 35, and 36, respectively; Figure 14). Compound 36, an adamantyl derivative, presented an antimalarial activity comparable to that of artemisinin and better than CQ against the Pf CQS D6, CQR W2 and multi-drug resistant TM91C235 strains (36: IC50(W2) = 8.6 nM, IC50(D6) = 9.3 nM, IC50(TM91C235) = 5.8 nM; Artemisinin: IC50(W2) = 6.70 nM, IC50(D6) = 9.00 nM, IC50(TM91C235) = 13.40 nM; CQ: IC50(W2) = 456.20 nM, IC50(D6) = 12.27 nM, IC50(TM91C235) = 138.82 nM). Compound cytotoxicity was evaluated in the human liver carcinoma cell line HepG2, and compound 36 exhibited the best selectivity index (SI(W2) = 352; SI(D6) = 326; SI(TM91C235) = 522) [42].
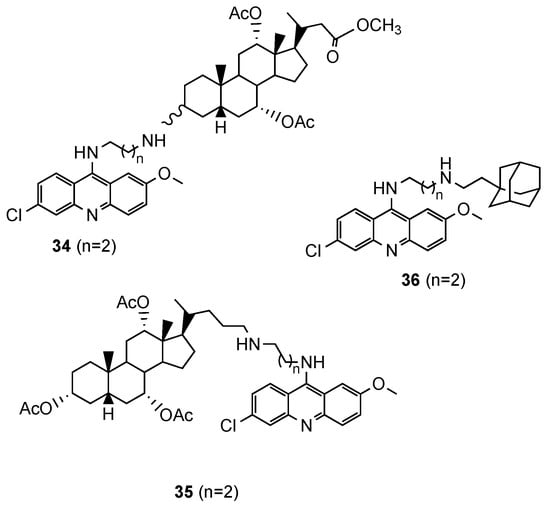
Figure 14. Steroidal (34 and 35) and adamantane (36) aminoacridine derivatives synthesized by Tot and co-workers [42].
Another approach was proposed by Dana et al. [43], who developed a novel class of hybrids integrating acridine and redox-active naphthalenediimide (NDI) scaffolds either directly linked (37a–e, Figure 15) or through a flexible linker (38a–c, Figure 15). Both series were further evaluated against Pf CQS 3D7 and CQR W2 strains. Overall, the compounds showed better activities than the parent drugs NDI and acridine (NDI: IC50 = 260 nM; Acridine: IC50 = 26.4 nM). The orthogonal series (37a–e) displayed considerably less antiplasmodial activity than the flexible series (38a–c), which demonstrated the influence of the spacer linker on the antimalarial activity (37a–e: 419 < IC50 < 4197 nM; 38a–c: 3.65 < IC50 < 8.44 nM). Moreover, structure–activity relationship studies proved once again the importance, for antiplasmodial activity, of the incorporation of the substituents 6-chloro and 2-methoxy in the acridine core. Hybrids 38a and 38b presented the best activities, alongside no cytotoxic in mammalian fibroblast NIH3T3 cells (38a: IC50(3D7) = 3.65 nM, IC50(W2) = 52.20 nM; 38b: IC50(3D7) = 4.33 nM, IC50(W2) = 28.53 nM; CQ: IC50(3D7) = 12.56 nM, IC50(W2) = 430.60 nM) [43].
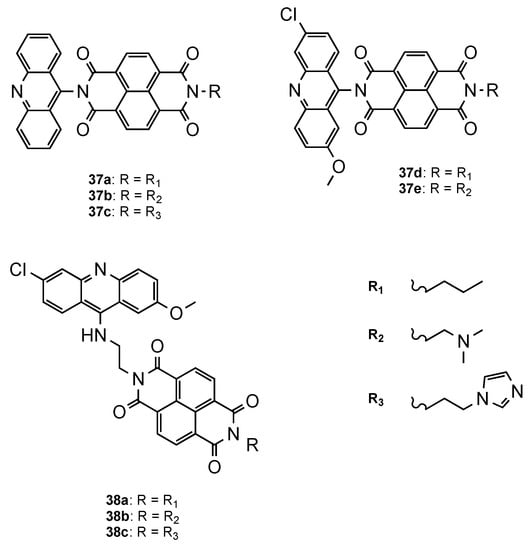
Figure 15. Orthogonal (37a–e) and flexible (38a–c) series of NDI–acridine hybrids developed by Dana and co-workers [43].
Considering the recent reports of resistance to ACT [3], Pandey et al. [44] developed a series of pyrrolidinoaminoalkane–acridine hybrids (39, Figure 16) to posteriorly use them as potential partners with artemisinin derivatives for the ACT. The two points of structural variations were the phenyl ring and the alkane spacer. When evaluated against the CQS 3D7 and the CQR K1 strains, compound 39a performed better (39a: IC50 (3D7) = 9.3 nM, IC50 (K1) = 5.2 nM). Then, 39a was evaluated in vivo in Swiss mice infected with the MDR strain P. yoelii nigeriensis at a dose of 100 mg/kg per day for 4 days via the oral route, showing > 90% suppression of parasitaemia on day 4. The authors further showed that, when tested in combination with artemether, arteether or artesunate, compound 39a serves as a good partner in ACT, namely, with artemether [44].
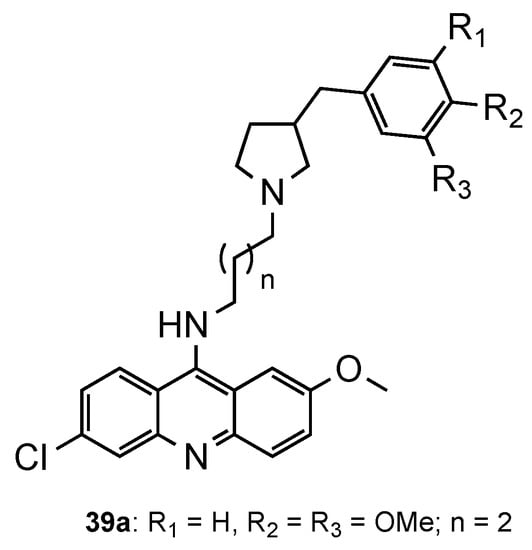
Figure 16. Most active compound 39a developed by Pandey and co-workers [44].
Recently, Dana et al. [45] focused their work on ciprofloxacin–acridine hybrids. Ciprofloxacin (CFX) is a second-generation fluoroquinolone-based antibiotic, known to be active against the in vitro culture of Pf [46][47]. In this work, the authors functionalized CFX in the N-terminal of the piperazine moiety with acridine scaffold. Hybrid 40 (Figure 17) was tested for its in vitro antimalarial activity against the CQS 3D7 strain of Pf. Although the activity of 40 was improved in comparison to that of CFX, it was not better than the reference CQ (3D7: IC50 (40) = 359.40 nM, IC50 (CFX) = 45350.0 nM, IC50 (CQ) = 12.56 nM). However, it is noteworthy that compound 40 did not show any hemolysis of infected RBCs or uninfected RBCs at their highest tested concentrations (10–45 µM), which may suggest minimal toxic effects in vivo [45].
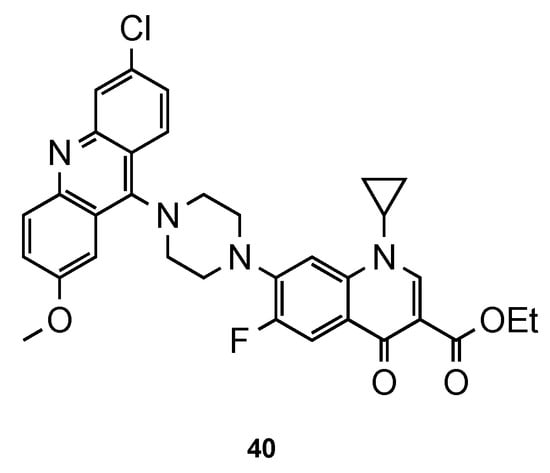
Figure 17. CFX–acridine hybrid 40 developed by Dana and co-workers [45].
Blackie et al. [48] established the role of ferrocenyl moiety in the antiplasmodial activity of ferroquine, another import scaffold in antimalarial derivatives [49]. The authors concluded that ferrocenyl fragment serves simply as a hydrophobic spacer group. Next, they synthesized compound 41 (Figure 18) and tested them against CQS 3D7 and CQR K1 strains of Pf. It is noteworthy that hybrid 41 showed potent antimalarial activity (ED50 = 1 and 8 nM against CQS 3D7 and CQR K1 Pf strains, respectively), comparatively to CQ (ED50 = 8.52 and 290 nM against CQS 3D7 and CQR K1 Pf strains, respectively) [48].
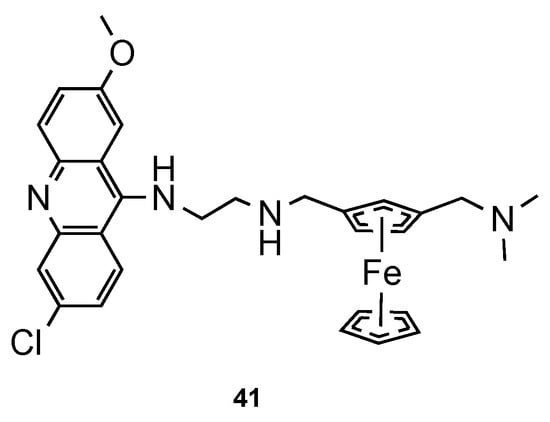
Figure 18. Ferrocene–acridine hybrid 41 developed by Blackie and co-workers [48].
This review depicts a brief background of the development of antimalarials based on the heteroaromatic core of QN focused on the last two decades. Despite the potent antimalarial activity of QN, its toxicity was among the reasons for its rapid substitution by CQ. Thus, all the modifications described in this review were mainly driven to increase the antimalarial activity of QN derivatives but also to improve their pharmacokinetic and pharmacodynamic properties, as well as overcome problems of cytotoxicity. The classical modifications in the side-chain of acridine reported in this paper emphasized the importance of the substituents methoxy and chlorine at positions 2 and 6 of the acridine ring, respectively. Still, it was also clear that small changes in the chemical structure of the molecules cause major impacts in their antimalarial activity. Alongside these modifications, this review also summarized the advances made towards hybrid molecules holding the acridine moiety. By joining two different molecules with a distinct mechanism of action, it is possible not only to improve the antiplasmodial activity but also to overcome resistance problems, often associated with classical antimalarial compounds. Although this approach does not always fulfill its purpose, it highlights the importance of the nature of the linker between the two moieties. It is noteworthy that the reported efforts have been progressively unveiling novel acridine-based derivatives as promising leads that will hopefully guide future research on the development of acridine-related molecules for the treatment of malaria.
References
- Cox, F.E. History of the discovery of the malaria parasites and their vectors. Parasit Vectors 2010, 3, 5.
- World Malaria Report 2019, World Health Organization. Available online: (accessed on 10 December 2020).
- World Health Organization. Artemisinin Resistance and Artemisinin-Based Combination Therapy Efficacy: Status Report (2018). Available online: (accessed on 10 December 2020.).
- Teixeira, C.; Vale, N.; Pérez, B.; Gomes, A.; Gomes, J.R.B.; Gomes, P. “Recycling” classical drugs for malaria. Chem. Rev. 2014, 114, 11164–11220.
- Delves, M.; Plouffe, D.; Scheurer, C.; Meister, S.; Wittlin, S.; Winzeler, E.A.; Sinden, R.E.; Leroy, D. The activities of current antimalarial drugs on the life cycle stages of plasmodium: A comparative study with human and rodent parasites. PLoS Med. 2012, 9, e1001169.
- Gensicka-Kowalewska, M.; Cholewiński, G.; Dzierzbicka, K. Recent developments in the synthesis and biological activity of acridine/acridone analogues. RSC Adv. 2017, 7, 15776–15804.
- Prasher, P.; Sharma, M. Medicinal chemistry of acridine and its analogues. MedChemComm 2018, 9, 1589–1618.
- Ehsanian, R.; Van Waes, C.; Feller, S.M. Beyond DNA binding–a review of the potential mechanisms mediating quinacrine’s therapeutic activities in parasitic infections, inflammation, and cancers. Cell Commun. Signal. 2011, 9, 13.
- Joanny, F.; Held, J.; Mordmuller, B. In vitro activity of fluorescent dyes against asexual blood stages of plasmodium falciparum. Antimicrob. Agents Chemother. 2012, 56, 5982–5985.
- Wainwright, M. Dyes in the development of drugs and pharmaceuticals. Dyes Pigments 2008, 76, 582–589.
- Kitchen, L.W.; Vaughn, D.W.; Skillman, D.R. Role of us military research programs in the development of us food and drug administration--approved antimalarial drugs. Clin. Infect. Dis. 2006, 43, 67–71.
- Meunier, B. Hybrid molecules with a dual mode of action: Dream or reality? Acc. Chem. Res. 2008, 41, 69–77.
- Agarwal, D.; Gupta, R.D.; Awasthi, S.K. Are antimalarial hybrid molecules a close reality or a distant dream? Antimicrob. Agents Chemother. 2017, 61.
- King, H.D.; Wilson, W.D.; Gabbay, E.J. Interactions of some novel amide-linked bis(acridines) with deoxyribonucleic acid. Biochemistry 1982, 21, 4982–4989.
- Girault, S.; Grellier, P.; Berecibar, A.; Maes, L.; Mouray, E.; Lemière, P.; Debreu, M.A.; Davioud-Charvet, E.; Sergheraert, C. Antimalarial, antitrypanosomal, and antileishmanial activities and cytotoxicity of bis(9-amino-6-chloro-2-methoxyacridines): Influence of the linker. J. Med. Chem. 2000, 43, 2646–2654.
- Girault, S.; Delarue, S.; Grellier, P.; Berecibar, A.; Maes, L.; Quirijnen, L.; Lemiere, P.; Debreu-Fontaine, M.A.; Sergheraert, C. Antimalarial in-vivo activity of bis(9-amino-6-chloro-2-methoxyacridines). J. Pharm. Pharmacol. 2001, 53, 935–938.
- Caffrey, C.R.; Steverding, D.; Swenerton, R.K.; Kelly, B.; Walshe, D.; Debnath, A.; Zhou, Y.-M.; Doyle, P.S.; Fafarman, A.T.; Zorn, J.A.; et al. Bis-acridines as lead antiparasitic agents: Structure-activity analysis of a discrete compound library in vitro. Antimicrob. Agents Chemother. 2007, 51, 2164–2172.
- Crespo-Ortiz, M.P.; Wei, M.Q. Antitumor activity of artemisinin and its derivatives: From a well-known antimalarial agent to a potential anticancer drug. J. Biomed. Biotechnol. 2012, 2012, 247597.
- Li, Y. Qinghaosu (artemisinin): Chemistry and pharmacology. Acta Pharmacol. Sin. 2012, 33, 1141–1146.
- O’Neill, P.M.; Posner, G.H. A medicinal chemistry perspective on artemisinin and related endoperoxides. J. Med. Chem. 2004, 47, 2945–2964.
- Araújo, N.C.P.; Barton, V.; Jones, M.; Stocks, P.A.; Ward, S.A.; Davies, J.; Bray, P.G.; Shone, A.E.; Cristiano, M.L.S.; O’Neill, P.M. Semi-synthetic and synthetic 1,2,4-trioxaquines and 1,2,4-trioxolaquines: Synthesis, preliminary sar and comparison with acridine endoperoxide conjugates. Bioorg. Med. Chem. Lett. 2009, 19, 2038–2043.
- Jones, M.; Mercer, A.E.; Stocks, P.A.; La Pensée, L.J.I.; Cosstick, R.; Park, B.K.; Kennedy, M.E.; Piantanida, I.; Ward, S.A.; Davies, J.; et al. Antitumour and antimalarial activity of artemisinin–acridine hybrids. Bioorg. Med. Chem. Lett. 2009, 19, 2033–2037.
- Joubert, J.P.; Smit, F.J.; du Plessis, L.; Smith, P.J.; N’Da, D.D. Synthesis and in vitro biological evaluation of aminoacridines and artemisinin-acridine hybrids. Eur. J. Pharm. Sci. 2014, 56, 16–27.
- Raynes, K.; Foley, M.; Tilley, L.; Deady, L.W. Novel bisquinoline antimalarials: Synthesis, antimalarial activity, and inhibition of haem polymerisation. Biochem. Pharmacol. 1996, 52, 551–559.
- Vennerstrom, J.L.; Ager, A.L.; Dorn, A.; Andersen, S.L.; Gerena, L.; Ridley, R.G.; Milhous, W.K. Bisquinolines. 2. Antimalarial n,n-bis(7-chloroquinolin-4-yl)heteroalkanediamines. J. Med. Chem. 1998, 41, 4360–4364.
- Liebman, K.M.; Burgess, S.J.; Gunsaru, B.; Kelly, J.X.; Li, Y.; Morrill, W.; Liebman, M.C.; Peyton, D.H. Unsymmetrical bisquinolines with high potency against p. Falciparum malaria. Molecules 2020, 25, 2251.
- Kumar, A.; Srivastava, K.; Kumar, S.R.; Puri, S.K.; Chauhan, P.M. Synthesis of new 4-aminoquinolines and quinoline-acridine hybrids as antimalarial agents. Bioorg. Med. Chem. Lett. 2010, 20, 7059–7063.
- Gutteridge, C.E.; Vo, J.V.; Tillett, C.B.; Vigilante, J.A.; Dettmer, J.R.; Patterson, S.L.; Werbovetz, K.A.; Capers, J.; Nichols, D.A.; Bhattacharjee, A.K.; et al. Antileishmanial and antimalarial chalcones: Synthesis, efficacy and cytotoxicity of pyridinyl and naphthalenyl analogs. J. Med. Chem. 2007, 3, 115–119.
- Li, R.; Kenyon, G.L.; Cohen, F.E.; Chen, X.; Gong, B.; Dominguez, J.N.; Davidson, E.; Kurzban, G.; Miller, R.E.; Nuzum, E.O.; et al. In vitro antimalarial activity of chalcones and their derivatives. J. Med. Chem. 1995, 38, 5031–5037.
- Tomar, V.; Bhattacharjee, G.; Kamaluddin; Rajakumar, S.; Srivastava, K.; Puri, S.K. Synthesis of new chalcone derivatives containing acridinyl moiety with potential antimalarial activity. Eur. J. Med. Chem. 2010, 45, 745–751.
- Prajapati, S.P.; Kaushik, N.K.; Zaveri, M.; Mohanakrishanan, D.; Kawathekar, N.; Sahal, D. Synthesis, characterization and antimalarial evaluation of new β-benzoylstyrene derivatives of acridine. Arab. J. Chem. 2017, 10, S274–S280.
- Pérez, B.; Teixeira, C.; Albuquerque, I.S.; Gut, J.; Rosenthal, P.J.; Prudêncio, M.; Gomes, P. Primacins, n-cinnamoyl-primaquine conjugates, with improved liver-stage antimalarial activity. MedChemComm 2012, 3, 1170–1172.
- Pérez, B.; Teixeira, C.; Gut, J.; Rosenthal, P.J.; Gomes, J.R.; Gomes, P. Cinnamic acid/chloroquinoline conjugates as potent agents against chloroquine-resistant plasmodium falciparum. ChemMedChem 2012, 7, 1537–1540.
- Pérez, B.C.; Teixeira, C.; Albuquerque, I.S.; Gut, J.; Rosenthal, P.J.; Gomes, J.R.B.; Prudêncio, M.; Gomes, P. N-cinnamoylated chloroquine analogues as dual-stage antimalarial leads. J. Med. Chem. 2013, 56, 556–567.
- Pérez, B.; Teixeira, C.; Gomes, A.S.; Albuquerque, I.S.; Gut, J.; Rosenthal, P.J.; Prudêncio, M.; Gomes, P. In vitro efficiency of 9-(n-cinnamoylbutyl)aminoacridines against blood- and liver-stage malaria parasites. Bioorg. Med. Chem. Lett. 2013, 23, 610–613.
- Gomes, A.; Pérez, B.; Albuquerque, I.; Machado, M.; Prudêncio, M.; Nogueira, F.; Teixeira, C.; Gomes, P. N-cinnamoylation of antimalarial classics: Quinacrine analogues with decreased toxicity and dual-stage activity. ChemMedChem 2014, 9, 305–310.
- Gemma, S.; Campiani, G.; Butini, S.; Kukreja, G.; Coccone, S.S.; Joshi, B.P.; Persico, M.; Nacci, V.; Fiorini, I.; Novellino, E.; et al. Clotrimazole scaffold as an innovative pharmacophore towards potent antimalarial agents: Design, synthesis, and biological and structure–activity relationship studies. J. Med. Chem. 2008, 51, 1278–1294.
- Gemma, S.; Campiani, G.; Butini, S.; Kukreja, G.; Joshi, B.P.; Persico, M.; Catalanotti, B.; Novellino, E.; Fattorusso, E.; Nacci, V.; et al. Design and synthesis of potent antimalarial agents based on clotrimazole scaffold: Exploring an innovative pharmacophore. J. Med. Chem. 2007, 50, 595–598.
- Gemma, S.; Campiani, G.; Butini, S.; Joshi, B.P.; Kukreja, G.; Coccone, S.S.; Bernetti, M.; Persico, M.; Nacci, V.; Fiorini, I.; et al. Combining 4-aminoquinoline- and clotrimazole-based pharmacophores toward innovative and potent hybrid antimalarials. J. Med. Chem. 2009, 52, 502–513.
- Kumar, A.; Srivastava, K.; Raja Kumar, S.; Puri, S.K.; Chauhan, P.M.S. Synthesis of 9-anilinoacridine triazines as new class of hybrid antimalarial agents. Bioorg. Med. Chem. Lett. 2009, 19, 6996–6999.
- Solaja, B.A.; Opsenica, D.; Smith, K.S.; Milhous, W.K.; Terzić, N.; Opsenica, I.; Burnett, J.C.; Nuss, J.; Gussio, R.; Bavari, S. Novel 4-aminoquinolines active against chloroquine-resistant and sensitive p. Falciparum strains that also inhibit botulinum serotype a. J. Med. Chem. 2008, 51, 4388–4391.
- Tot, M.; Opsenica, D.; Mitric, M.; Burnett, J.; Gomba, L.; Bavari, S.; Solaja, B. New 9-aminoacridine derivatives as inhibitors of botulinum neurotoxins and P. falciparum malaria. J. Serb. Chem. Soc. 2013, 78, 1847–1864.
- Dana, S.; Keshri, S.K.; Shukla, J.; Vikramdeo, K.S.; Mondal, N.; Mukhopadhyay, P.; Dhar, S.K. Design, synthesis and evaluation of bifunctional acridinine−naphthalenediimide redox-active conjugates as antimalarials. ACS Omega 2016, 1, 318–333.
- Pandey, S.K.; Biswas, S.; Gunjan, S.; Chauhan, B.S.; Singh, S.K.; Srivastava, K.; Singh, S.; Batra, S.; Tripathi, R. Pyrrolidine-acridine hybrid in artemisinin-based combination: A pharmacodynamic study. Parasitology 2016, 143, 1421–1432.
- Dana, S.; Valissery, P.; Kumar, S.; Gurung, S.K.; Mondal, N.; Dhar, S.K.; Mukhopadhyay, P. Synthesis of novel ciprofloxacin-based hybrid molecules toward potent antimalarial activity. ACS Med. Chem. Lett. 2020, 11, 1450–1456.
- Divo, A.A.; Sartorelli, A.C.; Patton, C.L.; Bia, F.J. Activity of fluoroquinolone antibiotics against plasmodium falciparum in vitro. Antimicrob. Agents Chemother. 1988, 32, 1182–1186.
- Dubar, F.; Anquetin, G.; Pradines, B.; Dive, D.; Khalife, J.; Biot, C. Enhancement of the antimalarial activity of ciprofloxacin using a double prodrug/bioorganometallic approach. J. Med. Chem. 2009, 52, 7954–7957.
- Blackie, M.A.L.; Beagley, P.; Croft, S.L.; Kendrick, H.; Moss, J.R.; Chibale, K. Metallocene-based antimalarials: An exploration into the influence of the ferrocenyl moiety on in vitro antimalarial activity in chloroquine-sensitive and chloroquine-resistant strains of plasmodium falciparum. Bioorg. Med. Chem. 2007, 15, 6510–6516.
- Peter, S.; Aderibigbe, B.A. Ferrocene-based compounds with antimalaria/anticancer activity. Molecules 2019, 24, 3604.


