 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Natalia Ptaszyńska | + 2554 word(s) | 2554 | 2021-02-07 04:39:42 | | | |
| 2 | Peter Tang | Meta information modification | 2554 | 2021-02-23 14:33:49 | | | | |
| 3 | Aleksandra Hawryłkiewicz | + 1336 word(s) | 3890 | 2021-02-24 10:42:37 | | | | |
| 4 | Peter Tang | + 1336 word(s) | 3890 | 2021-02-24 10:53:22 | | |
Video Upload Options
Gemcitabine is an anticancer drug used to treat a wide range of solid tumors and is a first-line treatment for pancreatic cancer. The effect of gemcitabine is significantly weakened by its rapid plasma degradation. In addition, the systemic toxicity and drug resistance significantly reduce its chemotherapeutic efficacy. Up to now, many approaches have been made to improve the therapeutic index of gemcitabine. One of the recently developed approaches to improve conventional chemotherapy is based on the direct targeting of chemotherapeutics to cancer cells using the peptide-drug conjugates (PDCs).
1. Introduction
Improving the therapeutic index of anticancer agents is an enormous challenge. In a time when the number of patients suffering from a cancer-related disease has been increasing each day and when conventional therapies gather a worrying number of deficits and drawbacks [1][2], new treatment options are required to relieve the symptoms and ultimately to eradicate the disease. Over the past decades, the development of new therapies that are more selective and less harmful to patients has been the target of many research groups [3][4]. However, these therapies still carry a risk of relapse and numerous side effects. The chemotherapeutic agents that target rapidly dividing cancer cells significantly damage healthy cells, especially those with rapid growth, such as bone marrow, gastrointestinal mucosa, and hair cells [5][6]. This causes the most common side effects of chemotherapy, such as myelosuppression (reduced blood cell production), inflammation of the lining of the gastrointestinal tract, and hair loss [6].
A modern approach to improve conventional chemotherapy is to focus on the direct targeting of chemotherapeutic agents to cancer cells [3][5]. Targeted drug delivery methods have been developed to improve drug efficacy and lower side effects by directing the drug to a specific cell type, enhance the tumoricidal effect, and reduce the peripheral toxicity of a specific drug [7]. Targeting decreases the side effects of therapeutic agents by delivering drugs to the intended destination [8]. To optimize the strategy of the anti-cancer agents targeting, one must ensure that the drug will not affect non-tumor-transformed cells and that the active substances will be transported in sufficient amounts to eliminate cancer cells and inhibit the tumor growth [8][9].
Designing the carriers of therapeutic substances is an approach that enables not only the improvement of pharmacokinetic properties and the biodistribution of traditional and innovative drugs, but also a reduction of their numerous side effects [9]. The binding of a drug to a carrier is often accompanied by a change in its mode of delivery, which is advantageous if it leads to the increased accumulation of the drug in the target tissue, e.g., in a cancer cell [5][8]. The biodistribution of such a conjugate is highly dependent on the properties of the carrier [10]. The synthesis of drug–peptide conjugates for the targeted delivery to a specific group of cells usually involves the conjugation of the drug with a targeting peptide via an appropriate linker, which could in turn facilitate the chemical or enzymatic release of the drug once the conjugate enters cancer cells via a receptor–mediated endocytosis mechanism [8][11]. The ester linkages, among several functional groups employed to connect the drug to the linker, have been widely utilized due to its possible release via an enzymatic (i.e., esterase–based) hydrolysis of the ester bond [12].
Several peptide-drug conjugates (PDCs) have been developed for different cancer types [13][14][15][16][17]. Some of these endeavors proceeded to clinical rating [16][17], while other provided crucial information about approaches that could enhance the stability of these molecules in circulation, thus improving their efficacy and reducing any associated toxicity [18].
Gemcitabine (2′,2′-difluorodeoxycytidine) (dFdC, Gem-used in the conjugate’s names)—a pyrimidine antimetabolite—is a prodrug with a demonstrated efficacy against a wide variety of cancers [19][20][21] and has been approved for use against colon, non-small cell lung, breast, pancreatic, bladder, and ovarian cancers [22][23]. Gemcitabine is transported into cells by nucleoside transporters: Human equilibrative (hENT1, hENT2) and concentrative nucleoside transporters (hCNT, hCNT2, and hCNT3) [24]. The drug molecule is then activated by deoxycytidine kinase (DCK)—a rate-limiting step for its pharmacological action—and phosphorylated to difluorodeoxycytidinemonophosphate (dFdCMP), which is further phosphorylated by the phosphate kinase enzyme into diphosphate (dFdCDP) and triphosphate (dFdCTP) forms. Both the dFdCDP and dFdCTP active forms show antitumor activity by the inhibition of the cellular DNA synthesis. dFdCTP incorporates into the DNA leading strand inhibiting DNA synthesis and subsequently leading to cell apoptosis. dFdCDP also has an indirect cytotoxic effect caused by the inhibition of ribonucleotide reductase. Finally, dFdC is rapidly metabolized into its inactive metabolite—2′,2′-difluoro-2′-deoxyuridine (dFdU)—by cytidine deaminase present in the blood, liver, and kidneys and is excreted through urine [25] (Figure 1).
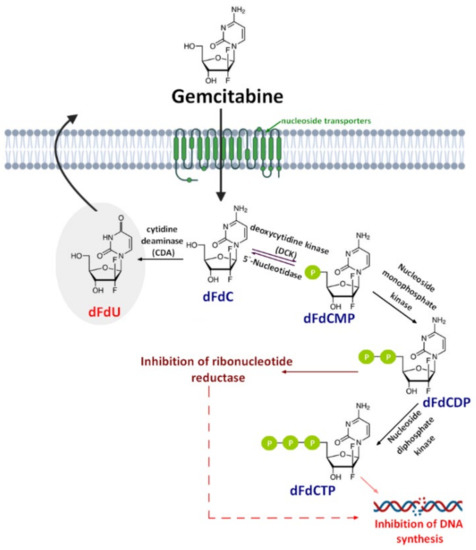
Figure 1. A schematic of gemcitabine (dFdC) cellular uptake, mechanism of action, and metabolism. dFdCMP: Gemcitabine monophosphate; dFdCDP: Gemcitabine diphosphate; dFdCTP: Gemcitabine triphosphate; and dFdU: 2′,2′-difluoro-2′-deoxyuridine.
Despite the clinical successful application of gemcitabine, its short plasma half-life (9−13 min in human plasma) [26], poor diffusion into cells, and adverse toxicity—such as myelosuppression, the principal dose-limiting toxicities in gemcitabine cancer therapy—significantly reduce its chemotherapeutic potential [27][28]. This short half-life is the result of deamination of gemcitabine by cytidine deaminase which—as mentioned before—metabolize gemcitabine to the inactive dFdU [29]. Likewise, phosphorylated metabolites of gemcitabine are inactivated via the reduction by cellular 5′-nucleotidase (5′-NT) and then are rapidly removed from the body by the enzymatic conversion of gemcitabine [30][31]. Another important drawback associated with gemcitabine therapy is the drug resistance related to the nucleoside transporter deficiency, which is developed by some tumor cells after the initial tumor regression [32]. For this reason, many approaches have been made to improve the safety profile of gemcitabine and increase its chemotherapeutic index. These approaches include both chemical modifications either on the cytosine’s aniline or on the 5′-hydroxyl group of the 2,2′-difluoro-2′-deoxyribose moiety [33] and the novel drug delivery technology. Until now, various delivery strategies such as liposomes [34][35][36], nanoparticles [37][38], lipidic and nonlipidic derivatives [36], as well as poly(ethylene glycol) (PEG) and other polymeric drug conjugates [39][40] have been studied to prevent rapid plasma degradation and improve the selective delivery of gemcitabine to the tumor tissue. These approaches have been widely discussed earlier [25] and will not be discussed again herein.
An alternative strategy, which has recently attracted much more attention, was established by chemically conjugating hydrophilic drugs to cell-penetrating peptides (CPPs) [41][42]. CPPs are relatively short peptides, typically composed of less than 30 amino acid, that have been shown to be comparatively non-cytotoxic and capable of crossing the cell membrane. These peptides have been used to facilitate the transport of various therapeutic agents into cells, including plasmid DNA, siRNA, therapeutic proteins, viruses, imaging agents, and other various nanoparticles. The coupling of the anticancer drug to CPPs may result in numerous advantages, such as improved solubility, intracellular uptake, biodistribution, and pharmacokinetic profiles. Therefore, the CPP-based drug delivery system offers great potential for improving the intracellular delivery of therapeutic agents with poor permeability [42].
2. Gemcitabine Conjugated with Cell-Penetrating Peptides (CPPs)
In recent years, numerous natural and synthetic CPPs—such as TAT, transportan, penetratin peptides, and polyamino acids (e.g., poly-arginines)—were utilized for the intracellular delivery of anticancer agents [43][44]. Since all CPPs are able to efficiently pass through cell membranes while being non-cytotoxic and carry a wide variety of cargos inside cells, they are also used to form conjugates with gemcitabine.
Vale et al. [41] synthesized two novel peptide–gemcitabine conjugates using two well-known CPP sequences—Penetratin (Pen, RQIKIWFQNRRMKWKK) [45] and pVEC (LLIILRRRIRKQAHAHSK) [46]. The authors connected these peptides with the aniline moiety of gemcitabine through the 3-sulfanylpropanoyl linker. They added an additional cysteine residue to the N-terminus of both CPPs to couple them with the linker, receiving Cys-Pen and Cys-pVec. A disulfide exchange reaction between Cys-modified CPPs and gemcitabine connected with the linker produced target conjugates (Figure 2B). The time-dependent kinetics of the gemcitabine release from hydrolysis of these new conjugates was studied in phosphate-buffered saline (PBS) at pH 7.4, 37 °C, and their biological activity was evaluated using three human cancer cell lines—MKN-28 (human gastric cancer), Caco-2 (heterogeneous human epithelial colorectal adenocarcinoma), and HT-29 (human colon adenocarcinoma). The results showed an increase in the anti-proliferative activity of gemcitabine in vitro upon conjugation with the CPP. Both CPP–gemcitabine conjugates (with the only exception of the Gem-Cys-pVEC conjugate against Caco-2 cells) worked substantially better than their components alone (either as a free drug or Cys-CPP) on the tested cell line (IC50 < 50 µM for conjugates vs. IC50 > 100 µM for gemcitabine and Cys-CPP). A semi-quantitative study of the degradation kinetics of both conjugates in PBS at pH 7.4, 37 °C, showed that gemcitabine is released upon the hydrolytic cleavage of the aromatic amide in Gem-Cys-Pen and Gem-Cys-pVec conjugates, with half-lives of approximately 9.6 days and 42 h, respectively. Based on the results mentioned above, the Vale’s group suggests that the remarkably higher stability of this conjugate may underlie its ability to make full use of its CPP moiety for enhanced internalization into the target cells, with a more controlled release of the parent drug over time [41].
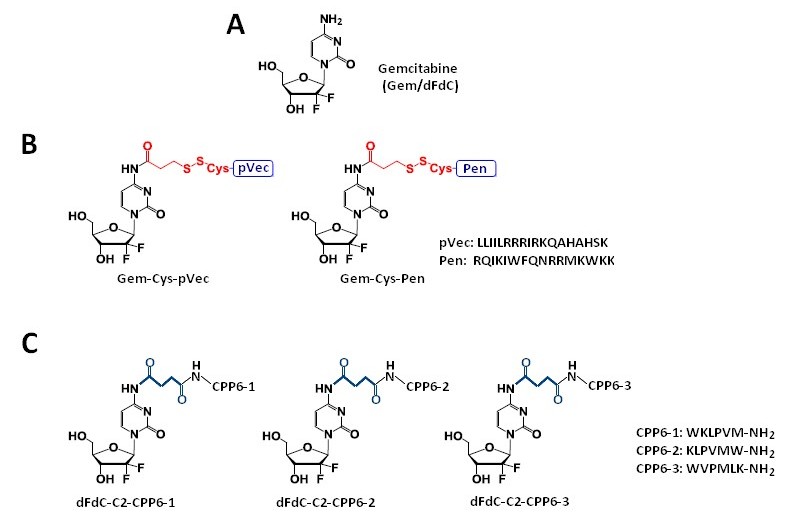
Figure 2. Chemical structures of gemcitabine (A) and their conjugates with CPPs and their constituents. (B) Gem-Cys-Pen and Gem-Cys-pVec conjugates: Conjugates of gemcitabine with Penetratin and pVec; (C) CPP6–dFdC: Conjugates of gemcitabine with CPP6.
In order to study the pharmacokinetics of gemcitabine-CPP conjugates and its constituents (gemcitabine and respective CPPs) and to establish a possible relation between the penetration potency of CPP and their physicochemical properties, Ferreira et al. [47] used the computational tool GastroPlus™—a powerful mechanistically-based simulation and modeling software for pharmaceutical research. Based on the simulations carried out in GastroPlus™, the authors stated that the conjugates’ bioavailability is ensured and the plasma concentration should reach therapeutic levels. The calculated AUC (area under the plasma concentration–time curve, μg-h/mL) for the conjugates was comparable to the AUC calculated for gemcitabine (~7.4404 and 7.4368, respectively). Yet, the estimated Cmax (maximum plasma concentration, in μg/mL) was higher for all the peptides and the analyzed conjugates (Cmax = 7.4403 μg/mL) compared with gemcitabine alone (Cmax = 5.9505 μg/mL). The Gem-Cys-pVEC conjugate binds less extensively to plasma proteins (>Fup, 42.89%). Bearing in mind that this conjugate showed the best in vitro bioactivity result for MKN-28 and HT-29 cells (IC50 = 20.68 µM and 45.20 µM, respectively; IC50 > 100 µM for gemcitabine) and released gemcitabine in PBS faster than the Gem-Cys-Pen conjugate (50% over 42 h versus 9.6 days for Gem-Cys-Pen) [41], the authors suggested that Gem-Cys-pVec conjugate has the most suitable profile for the drug delivery [47].
Continuing the previous studies, Vale’s group synthesized new series of gemcitabine-CPPs conjugates [32] containing three novel hexapeptides—CPP6-1, CPP6-2, and CPP6-3—which are the analogues of two peptides—KLPVM and VPMLK—derived from a family of CPP5 [48]. These peptides were reported to have a high ability to cross cell membranes. To improve their cell penetration capacity, additional tryptophan residues at their N- or/and C-termini were added and linked with gemcitabine using succinic anhydride, resulting in three novel gemcitabine-CPP6 conjugates (Figure 2C). Such a linker should improve the rate of drug delivery, as it protects the drug from cytidine deaminase (CDA) due to the conversion of its amino group to an amide moiety, comparatively unreactive under physiological conditions. To evaluate the in vitro cytotoxicity of the synthesized conjugates, the authors used human pancreatic adenocarcinoma (BxPC-3), human breast adenocarcinoma (MCF-7), and human prostate adenocarcinoma (PC-3) cancer cell lines. The results showed that two of three synthesized conjugates (dFdC-C2-CPP6-1 and dFdC-C2-CPP6-3) displayed a significantly higher cell growth inhibitory activity against PC-3 cells as compared with gemcitabine and CPPs constituent (after the conjugation with CPP6-1 and CPP6-3, gemcitabine IC50 decreased from 74 nM to 15 nM and 14 nM, respectively). Moreover, the three new conjugates of gemcitabine with CPP6 presented more potent cell growth inhibitory activity in MCF-7 and PC-3 cells (IC50 < 7 nM and IC50 ≤ 15 nM, respectively) than the reference drugs, tamoxifen (IC50 = 20 nM for MCF-7 and IC50 > 1000 nM for PC-3 cells) or metformin (IC50 = 9.9 nM for MCF-7 and IC50 = 189 nM for PC-3 cells) with the exception of the dFdC-C2-CPP6-2 conjugate for the PC-3 cell line (IC50 > 1000 nM). In addition, during this study the authors confirmed that in BxPC3 cells the dFdC-CPP6 conjugates are transported preferentially by hENT-1 transporter, and that once in the cytoplasm, dFdC-CPP6 conjugates may undergo sequential phosphorylations, disrupting DNA synthesis and causing apoptosis [32].
3. RGD Peptides-Gemcitabine Conjugates
Synthetic peptides containing the RGD sequence constitute one of the major classes of new pharmaceuticals with integrins as their primary therapeutic target. Such peptides act by binding to integrin receptors and destroying the mitochondria after cell penetration. This affects angiogenesis, thus disrupting the process of creating new blood vessels and inhibiting the tumor growth.
Unmodified RGD-peptide (Arg-Gly-Asp) binds specifically to αVβ3 and αVβ5 receptors, such as fibrinogen, fibronectin, vitronectin, plasminogen, thrombospondin, prothrombin, MMP-2, laminin, osteopontin, etc. which are excessively expressed on tumor cells and surfaces of vasculature, and is applied as an important component of a delivery system of various agents: Anticancer drugs, nanoparticles, imaging compounds, and virus vectors to tumor or angiogenic vessels [49][50][51][52][53]. The affinity of RGD peptides for their molecular targets may be affected by their conformation [54][55]. Apart from the direct interactions between peptide’s functional groups and their receptors, the conformational properties of the RGD motif can also be modified. The cyclization is a common modification, improving the binding features of RGD peptides and the rigidity of the molecule. Linear RGD peptides are strongly susceptible to the enzymatic degradation, but the introduction of conformational constrains caused by the cyclization averts this process, so cyclic peptides are more stable, more potent, and more specific. In cyclic peptides, the RGD-peptide sequence is surrounded by other amino acid residues to build a ring system. These modification offers the possibility to present the RGD sequence in a specific conformation for a selected integrin [54][55].
The iRGD (C(&)RGDKGPDC(&)) is one of the most commonly used tumor-targeting peptides. This peptide binds to the alphaVbeta3 and alphaVbeta5 integrins and to neuropilin-1 (NRP1)receptors increasing the pore diameter and surface area of blood vessels in the tumor, reducing the influence of pressure in its interstitium, and, in consequence, increasing the diffusion rate of small molecule drugs [53]. Drugs, nanoparticles, or proteins conjugated with iRGD can be directly administered to the tumor parenchyma, which reduce the side effects of the chemotherapy. Due to this innovative delivery system and the lowtoxicity to normal cells, iRGD has drawn the attention of several research groups [51]. Coadministration of this peptide has been proven to significantly enhance the intratumoralaccumulation of various molecules, such as, doxorubicin (7-fold), dextran (3- to 5-fold), Evans blue dye (2- to 4-fold), trastuzumab (40-fold), doxorubicin-liposome (14-fold), and nab-paclitaxel (9- to 12-fold) in the mouse breast or prostate cancer models [56]. Akashi et al. [57] demonstrated that the co-administration of gemcitabine and iRGD peptide boost the anticancer effect of the therapy. Studies conducted on the five mouse pancreatic cancer cell xenograft models showed that the high expression of neuropilin-1 enhanced the anticancer effect of gemcitabine combined with iRGD in comparison with its monotherapy. They also proved that there is a significant difference in the overall survival between patients showing high NRP1 expression (0.5–46.6 months) and low NRP1 expression (5.6–94.6 months), which implies that co-administration of the toxic agent with iRGD peptide could be beneficial for cancer patients with NRP1-overexpressing tumors.
Temming et al. reported that the conjugation of a (&RGDfK&) peptide on the liposomes surface significantly improves the intracellular gemcitabine uptake in the human ovarian cancer cell line SKOV-3. Additionally, this conjugate is relativelyneutral towards human normal red blood cells (RBCs) [58]. Higher intracellular drugconcentration caused by (&RGDfK&) loaded liposomes may affect crucial processes in cancer cells, such as the reduction of the mitochondrial membrane potential or the increase in the reactive oxygen species (ROS) level, inducing the apoptosis in cancer cells [59][60]. Unlike the necrosis, the apoptosis is a genetically-controlled process which does not entail inflammation in the human body, thus most of the chemotherapeutic drugs are designed to induce this process [61]. Tang et al. [62] showed that PEGylated liposomes (LPs) consisting of RGD tripeptide and gemcitabine upregulate the pro-apoptotic Bax protein, downregulate the anti-apoptotic Bcl-2 protein expression, and increase the apoptosis related protein caspase-3 expression in SKOV3 cell lines in comparison to the free drug and gemcitabine LPs without RGD peptide. Moreover, they determined that the RGD-Gem-LPs conjugate improves pharmacokinetic properties of gemcitabine, such as the biological half-life (t1/2), the bioavailability (AUC), and the mean residence time (MRT) after intravenous administration to rats. Cai et al. [63] confirmed that the RGD coating of Gem-LPs results in a lower toxicity and a much higher efficacy in MDA-MB-231 bearing mice compared with non-modified drug liposomes. This coated nanocarriers also exhibited a more beneficial entrapment efficiency (EE), physical stability, particle size, and shape than the liposomal gemcitabine. Yu et al. [64] showed that the (&RGD&) peptide conjugation on the surface of gemcitabine albumin nanoparticles (Gem-HSA-NPs) could be beneficial for pancreatic cancer patients. The in vitro results conducted on the BxPC-3 cell line confirmed that the (&RGD&) anchored nanoparticles can deliver gemcitabine to cells more efficiently than the non-targeting (&RGD&)-conjugated nanoparticles. After conjugation with the (&RGD&) peptide, the cellular uptake of (&RGD&)-HSA-NPs increased 3-fold in comparison with that of HSA-NPs. These results confirmed that the coating of the particle surface with (&RGD&) significantly enhances the uptake of nanoparticles by BxPC- 3 cells. Moreover, when comparing with the free gemcitabine, 4-N-myristoyl-gemcitabine, and Gem-HSA-NPs, (&RGD&)-Gem-HSA-NPs exhibited a significantly higher apoptosis rate. This result was consistent with the cytotoxicity study and indicates that (&RGD&)-Gem-HSA-NPs induced stronger early and late apoptosis. This effect was caused by major gemcitabine intracellular uptake resulting from the active targeting by the (&RGD&) peptide giving higher cytotoxicity than the other compounds.
The paclitaxel/gemcitabine combination chemotherapy is one of the preferred treatments for metastatic breast cancer patients. The combination chemotherapy of paclitaxel and gemcitabine has survival benefits and a tolerable toxicity profile [65]. This combination strategy reduces the risk of insufferable side effects and maximizes the therapeutic effect, even at a lower dose [66][67][68]. Nevertheless, co-delivery of gemcitabine and paclitaxel without carriers is limited by various problems.These include, e.g., short t1/2, instability, and low cellular permeability of gemcitabine and the hydrophobic character of paclitaxel. To solve this problem, Zhang et al. [69] encapsulated gemcitabine monophosphate and paclitaxel in the (&RGDfC&)-modified nanoparticles. Studies conducted on MCF-7 tumor-bearing mice showed a prolongedelimination from the bloodstream and a higher maximum plasma concentration for paclitaxel and gemcitabine monophosphate (Gmp) in comparison with the free paclitaxel and gemcitabine (p < 0.05). Moreover, tumors treated with modified nanocarriers exhibit a decrease in the number of mitotic figures, more basophilic and uniform nuclei, and the reduction in the expression of Bcl-2 and Bcl-xL proteins. These studies also demonstrated that there is no significant difference between Ptx/Gmp-NPs and the untreated group in the levels of white blood cells, red blood cells, hematocrit, and hemoglobin. Despite the inevitable presence of the nanocarriers accumulated in the lungs or liver, their amount was significantly lower when comparing it with the tumor tissue. With the help of nanoparticles containing (&RGD&) as a targeting ligand, most of the nanoparticles are taken up by tumor cells.
Han et al. [70] designed and synthesized an innovative multifunctional gemcitabine prodrug TPE-Gem-RGD which can be used for the targeted intracellular light-up imaging and the selective release of gemcitabine under the reductive environment inside cells. This conjugate contains: (1) Gemcitabine as a chemotherapeutic drug, (2) tetraphenylene (TPE) for the cell imaging, (3) GFLG (glycyl-L-phenyloalanyl-L-lecylglycine) linker sensitive to cathepsin B [71], (4) five residues of Asp to improve solubility of the conjugate, and (5) RGD tripeptide as a targeting group (Figure 3). Their studies proved that the RGD-targeted TPE-Gem-RGD conjugate is more effective in the inhibition of the BxPC-3 pancreatic cancer cells proliferation in comparison with the gemcitabine and the non targeted TPE-Gem-RDG prodrug. This suggests that active targeting is a meaningful aspect of the anticancer drug design process. Moreover, the TPE-Gem-RGD prodrug is internalized by cells through RGD-mediated endocytosis, which makes this conjugate more effective in the suppression of BxPC-3 cells than TPE-Gem-RDG.
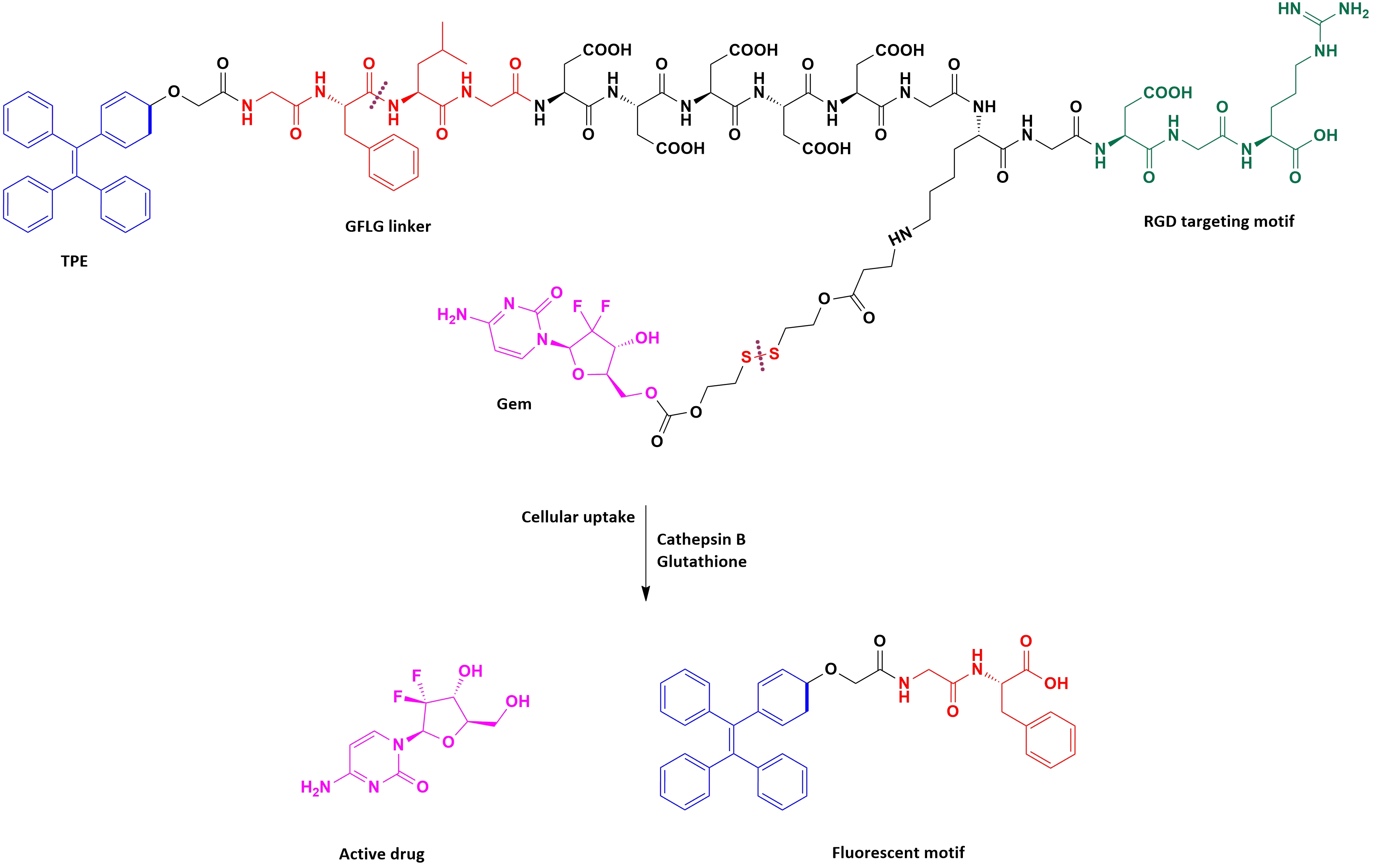
Figure 3. The structure of TPE-Gem-RGD for cathepsin B-responsive fluorescent product and reduction-responsive drug release. After cleavage of GFLG peptide by cathepsin B, intracellular fluorescence light up could be achieved. The conjugated anticancer drug- gemcitabine- is expected to be released upon the trigger of high intracellular glutathione concentration.
In order to prolong the half-life, overcome the drug resistance, and eliminate the bone marrow toxicity of gemcitabine, Liu et al. [72] designed and synthesized the RGDVgemcitabine conjugate. A lot of assays were carried out, including in vitro halflife assay, in vitro drug resistance assay, in vivo anti-tumor assay, in vivo kidney toxicity assay, in vivo liver toxicity assay, and in vivo marrow toxicity assay. Results, performed on a S180 tumor-bearing mouse model, indicated a 100-fold lower minimal effective dose, 10-fold higher anti-tumor activity, and 17-fold longer half-life (in mouse plasma) of the RGDVgemcitabine conjugate in comparison with gemcitabine alone. Moreover, based on FT(+)-MS spectrum analysis, RGDV-gemcitabine does not enter the liver, kidney, and marrow of the treated tumor-bearing mice. This means that the conjugate showed no kidney toxicity,no liver toxicity, and no marrow toxicity. To evaluate the drug resistance, the dipyridamole was used against the proliferation of A549 cells. The results showed that dipyridamole elevates the IC50 of gemcitabine from 2.5 µM to 48.0 µM (19.2-fold increase) but has little influence on the IC50 of RGDV-gemcitabine (elevation from 2.5 µM to 4.7 µM, only 1.9-fold increase). Thus, the modification of Arg-Gly-Asp-Val successively reverses dipyridamole-induced drug resistance. There is no significant differences between IC50 values for gemcitabine and RGDV-gemcitabine inhibiting the proliferation of MCF-7, HCT-8, A549, 95D, and HepG2 cells. Therefore, the Arg-Gly-Asp-Val modification does not change the in vitro anti-tumor activity of gemcitabine.
4. Conclusions
Drug targeting is crucial for effective cancer chemotherapy. Targeted delivery enhances chemotherapeutic effect and spares normal tissues from the toxic side effects of these powerful drugs. Up to now, many approaches have been made to improve the therapeutic index of gemcitabine. Peptide conjugates provide a valuable alternative to anti-cancer drugs used so far. A proper use of biological mechanisms of constituent peptide action can result in effective therapy for many diseases. Two groups of peptides were used to obtain gemcitabine conjugates—cell penetrating peptides and RGD peptides. Published results confirmed that peptides of both groups could be successfully applied to design new, efficient, and specific anti-cancer therapeutic agents, also in conjunction with the nanocarriers, such as nanoparticles, liposomes, or micelles. Enhanced pharmacological activity was achieved when components were non-covalently and covalently bond through the drug’s functional groups. The latter means that these groups could be subjected to the successful modification which usually is not the case for commercially available drugs. This again proves that gemcitabine is a very attractive leading structure to design gemcitabine conjugates with a potential to become new therapeutic tools for cancer therapy.
In conclusion, the use of gemcitabine peptide-based conjugates to treat cancer is a relatively new field and there are still many areas that need to be explored. However, these conjugates show great promise in the field of anticancer therapy because of their many benefits including no hematological or other toxicities, higher stability, and tissue specificity. One can expect that the successful transition from the laboratory to the clinic is only a matter of time and while the shift from the laboratory to the clinic is time consuming, and recent progress should promote this translation.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2018, 68, 394–424.
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer Incidence and Mortality Worldwide: Sources, Methods and Major Patterns in GLOBOCAN, 2012. Int. J. Cancer 2015, 136, 359–386.
- Duncan, R.; Vicent, M.J.; Greco, F.; Nicholson, R.I. Polymer-Drug Conjugates: Towards a Novel Approach for the Treatment of Endrocine-Related Cancer. Endocr. Relat. Cancer 2005, 12, 189–200.
- Allen, T.M. Ligand-Targeted Therapeutics in Anticancer Therapy. Nat. Rev. Cancer 2002, 2, 750–763.
- Allen, T.M.; Cullis, P.R. Drug Delivery Systems: Entering the Mainstream. Science 2004, 303, 1818–1822.
- Riedl, S.; Zweytick, D.; Lohner, K. Membrane-Active Host Defense Peptides—Challenges and Perspectives for the Development of Novel Anticancer Drugs. Chem. Phys. Lipids 2011, 164, 766–781.
- Hirabayashi, H.; Nishikawa, M.; Takakura, Y.; Hashida, M. Development and pharmacokinetics of galactosylated poly-L-glutamic acid as a biodegradable carrier for liver-specific drug delivery. Pharm Res. 1996, 13, 880–884.
- Böhme, D.; Beck-Sickinger, A.G. Drug Delivery and Release Systems for Targeted Tumor Therapy. J. Pept. Sci. 2015, 21, 186–200.
- Haag, R.; Kratz, F. Polymer Therapeutics: Concepts and Applications. Angew. Chem. Int. Ed. 2006, 45, 1198–1215.
- Nevozhay, D.; Kańska, U.; Budzyńska, R.; Boratyński, J. Current Status of Research on Conjugates and Related Drug Delivery Systems in the Treatment of Cancer and Other Diseases. Postepy Hig. Med. Dosw. 2007, 61, 350–360.
- Moulton, H.M.; Nelson, M.H.; Hatlevig, S.A.; Reddy, M.T.; Iversen, P.L. Cellular Uptake of Antisense Morpholino Oligomers Conjugated to Arginine-Rich Peptides. Bioconjug. Chem. 2004, 15, 290–299.
- Majumdar, S.; Siahaan, T.J. Peptide-Mediated Targeted Drug Delivery. Med. Res. Rev. 2012, 32, 637–658.
- Schreier, V.N.; Mezo, G.; Orbán, E.; Dürr, C.; Marquardt, A.; Manea, M. Synthesis, Enzymatic Stability and in Vitro Cytostatic Effect of Daunorubicin-GnRH-III Derivative Dimers. Bioorganic Med. Chem. Lett. 2013, 23, 2145–2150.
- Leurs, U.; Mezo, G.; Orbán, E.; Ohlschläger, P.; Marquardt, A.; Manea, M. Design, Synthesis, in Vitro Stability and Cytostatic Effect of Multifunctional Anticancer Drug-Bioconjugates Containing GnRH-III as a Targeting Moiety. Biopolymers 2012, 98, 1–10.
- Seitz, S.; Buchholz, S.; Schally, A.V.; Weber, F.; Klinkhammer-Schalke, M.; Inwald, E.C.; Perez, R.; Rick, F.G.; Szalontay, L.; Hohla, F.; et al. Triple Negative Breast Cancers Express Receptors for LHRH and Are Potential Therapeutic Targets for Cytotoxic LHRH-Analogs, AEZS 108 and AEZS 125. BMC Cancer 2014, 14, 847.
- Liu, S.V.; Tsao-Wei, D.D.; Xiong, S.; Groshen, S.; Dorff, T.B.; Quinn, D.I.; Tai, Y.C.; Engel, J.; Hawes, D.; Schally, A.V.; et al. Phase I, Dose-Escalation Study of the Targeted Cytotoxic Lhrh Analog Aezs-108 in Patients with Castration- and Taxane-Resistant Prostate Cancer. Clin. Cancer Res. 2014, 20, 6277–6283.
- Emons, G.; Gorchev, G.; Sehouli, J.; Wimberger, P.; Stähle, A.; Hanker, L.; Hilpert, F.; Sindermann, H.; Gründker, C.; Harter, P. Efficacy and Safety of AEZS-108 (INN: Zoptarelin Doxorubicin Acetate) an LHRH Agonist Linked to Doxorubicin in Women with Platinum Refractory or Resistant Ovarian Cancer Expressing LHRH Receptors: A Multicenter Phase II Trial of the Ago-Study Group (AGO G). Gynecol. Oncol. 2014, 133, 427–432.
- Karampelas, T.; Skavatsou, E.; Argyros, O.; Fokas, D.; Tamvakopoulos, C. Gemcitabine Based Peptide Conjugate with Improved Metabolic Properties and Dual Mode of Efficacy. Mol. Pharm. 2017, 14, 674–685.
- Ishikawa, T.; Kamimura, H.; Tsuchiya, A.; Togashi, T.; Watanabe, K.; Seki, K.; Ohta, H.; Yoshida, T.; Takeda, K.; Kamimura, T. Clinical Efficacy of Intra-Arterial Pharmacokinetic Chemotherapy with 5-Fluorouracil, CDDP, Gemcitabine, and Angiotensin-II in Patients with Advanced Pancreatic Cancer. Hepatogastroenterology 2007, 54, 2378–2382.
- Reddy, L.; Couvreur, P. Novel Approaches to Deliver Gemcitabine to Cancers. Curr. Pharm. Des. 2008, 14, 1124–1137.
- Lund, B.; Hansen, O.P.; Theilade, K.; Hansen, M.; Neijt, J.P. Phase II Study of Gemcitabine (2′,2′-Difluorodeoxycytidine) in Previously Treated Ovarian Cancer Patients. J. Natl. Cancer Inst. 1994, 86, 1530–1533.
- Plunkett, W.; Huang, P.; Gandhi, V. Preclinical Characteristics of Gemcitabine. Anticancer Drugs 1995, 6, 7–13.
- Hertel, L.W.; Boder, G.B.; Kroin, J.S.; Rinzel, S.M.; Poore, G.A.; Todd, G.C.; Grindey, G.B. Evaluation of the Antitumor Activity of Gemcitabine (2′,2′-Difluoro-2′-Deoxycytidine). Cancer Res. 1990, 50, 4417–4422.
- Mini, E.; Nobili, S.; Caciagli, B.; Landini, I.; Mazzei, T. Cellular Pharmacology of Gemcitabine. Ann. Oncol. 2006, 17, 7–12.
- Dyawanapelly, S.; Kumar, A.; Chourasia, M.K. Lessons Learned from Gemcitabine: Impact of Therapeutic Carrier Systems and Gemcitabine’s Drug Conjugates on Cancer Therapy. Crit. Rev. Ther. Drug Carrier Syst. 2017, 34, 63–69.
- Reid, J.M.; Qu, W.; Safgren, S.L.; Ames, M.M.; Krailo, M.D.; Seibel, N.L.; Kuttesch, J.; Holcenberg, J. Phase I Trial and Pharmacokinetics of Gemcitabine in Children with Advanced Solid Tumors. J. Clin. Oncol. 2004, 22, 2445–2451.
- Abbruzzese, J.L.; Grunewald, R.; Weeks, E.A.; Gravel, D.; Adams, T.; Nowak, B.; Mineishi, S.; Tarassoff, P.; Satterlee, W.; Raber, M.N. A phase I clinical, plasma, and cellular pharmacology study of gemcitabine. J. Clin. Oncol. 1991, 9, 491–498.
- Grunewald, R.; Kantarjian, H.; Du, M.; Faucher, K.; Tarassoff, P.; Plunkett, W. Gemcitabine in Leukemia: A Phase I Clinical, Plasma, and Cellular Pharmacology Study. J. Clin. Oncol. 1992, 10, 406–413.
- Immordino, M.L.; Brusa, P.; Rocco, F.; Arpicco, S.; Ceruti, M.; Cattel, L. Preparation, Characterization, Cytotoxicity and Pharmacokinetics of Liposomes Containing Lipophilic Gemcitabine Prodrugs. J. Control. Release 2004, 100, 331–346.
- Alvarellos, M.L.; Lamba, J.; Sangkuhl, K.; Thorn, C.F.; Wang, L.; Klein, D.J.; Altman, R.B.; Klein, T.E. PharmGKB Summary: Gemcitabine Pathway. Pharmacogenet. Genom. 2014, 24, 564–574.
- Kroep, J.; Van Moorsel, C.; Veerman, G.; Voorn, D.; Schultz, R.; Worzalla, J.; Tanzer, L.; Merriman, R.; Pinedo, H.; Peters, G. Role of Deoxycytidine Kinase (DCK), Thymidine Kinase 2 (TK2), and Deoxycytidine Deaminase (DCDA) in the Antitumor Activity of Gemcitabine (DFdC). In Purine and Pyrimidine Metabolism in Man IX; Springer: Boston, MA, USA, 1998; pp. 657–660.
- Correia, C.; Xavier, C.P.R.; Duarte, D.; Ferreira, A.; Moreira, S.; Vasconcelos, M.H.; Vale, N. Development of Potent CPP6-Gemcitabine Conjugates against Human Prostate Cancer Cell Line (PC-3). RSC Med. Chem. 2020, 11, 268–273.
- Moysan, E.; Bastiat, G.; Benoit, J.P. Gemcitabine versus Modified Gemcitabine: A Review of Several Promising Chemical Modifications. Mol. Pharm. 2013, 10, 430–444.
- May, J.P.; Ernsting, M.J.; Undzys, E.; Li, S.D. Thermosensitive Liposomes for the Delivery of Gemcitabine and Oxaliplatin to Tumors. Mol. Pharm. 2013, 10, 4499–4508.
- Grazia Calvagno, M.; Celia, C.; Paolino, D.; Cosco, D.; Iannone, M.; Castelli, F.; Doldo, P.; Fresta, M. Effects of Lipid Composition and Preparation Conditions on Physical-Chemical Properties, Technological Parameters and In Vitro Biological Activity of Gemcitabine-Loaded Liposomes. Curr. Drug Deliv. 2006, 4, 89–101.
- Bersani, S.; Vila-Caballer, M.; Brazzale, C.; Barattin, M.; Salmaso, S. PH-Sensitive Stearoyl-PEG-Poly(Methacryloyl Sulfadimethoxine) Decorated Liposomes for the Delivery of Gemcitabine to Cancer Cells. Eur. J. Pharm. Biopharm. 2014, 88, 670–682.
- Lee, G.Y.; Qian, W.P.; Wang, L.; Wang, Y.A.; Staley, C.A.; Satpathy, M.; Nie, S.; Mao, H.; Yang, L. Theranostic Nanoparticles with Controlled Release of Gemcitabine for Targeted Therapy and MRI of Pancreatic Cancer. ACS Nano 2013, 7, 2078–2089.
- Dolatabadi, J.E.N.; Valizadeh, H.; Hamishehkar, H. Solid Lipid Nanoparticles as Efficient Drug and Gene Delivery Systems: Recent Breakthroughs. Adv. Pharm. Bull. 2015, 5, 151–159.
- Aggarwal, S.; Gupta, S.; Pabla, D.; Murthy, R.S.R. Gemcitabine-Loaded PLGA-PEG Immunonanoparticles for Targeted Chemotherapy of Pancreatic Cancer. Cancer Nanotechnol. 2013, 4, 145–157.
- Chitkara, D.; Mittal, A.; Behrman, S.W.; Kumar, N.; Mahato, R.I. Self-Assembling, Amphiphilic Polymer-Gemcitabine Conjugate Shows Enhanced Antitumor Efficacy against Human Pancreatic Adenocarcinoma. Bioconjug. Chem. 2013, 24, 1161–1173.
- Vale, N.; Ferreira, A.; Fernandes, I.; Alves, C.; Araújo, M.J.; Mateus, N.; Gomes, P. Gemcitabine Anti-Proliferative Activity Significantly Enhanced upon Conjugation with Cell-Penetrating Peptides. Bioorganic Med. Chem. Lett. 2017, 27, 2898–2901.
- Zakeri-Milani, P.; Farkhani, S.M.; Shirani, A.; Mohammadi, S.; Mojarrad, J.S.; Akbari, J.; Valizadeh, H. Cellular Uptake and Anti-Tumor Activity of Gemcitabine Conjugated with New Amphiphilic Cell Penetrating Peptides. EXCLI J. 2017, 16, 650–662.
- Gupta, A.; Mandal, D.; Ahmadibeni, Y.; Parang, K.; Bothun, G. Hydrophobicity Drives the Cellular Uptake of Short Cationic Peptide Ligands. Eur. Biophys. J. 2011, 40, 727–736.
- Nasrolahi Shirazi, A.; Mandal, D.; Tiwari, R.K.; Guo, L.; Lu, W.; Parang, K. Cyclic Peptide-Capped Gold Nanoparticles as Drug Delivery Systems. Mol. Pharm. 2013, 10, 500–511.
- Dupont, E.; Prochiantz, J.A. Penetratin Story: An Overview. Methods Mol. Biol. 2015, 1324, 29–37.
- Nan, Y.H.; Park, I.S.; Hahm, K.S.; Shin, S.Y. Antimicrobial Activity, Bactericidal Mechanism and LPS-Neutralizing Activity of the Cell-Penetrating Peptide PVEC and Its Analogs. J. Pept. Sci. 2011, 17, 812–817.
- Ferreira, A.; Lapa, R.; Vale, N. Combination of Gemcitabine with Cell-Penetrating Peptides: A Pharmacokinetic Approach Using in Silico Tools. Biomolecules 2019, 9, 693.
- Gomez, J.A.; Chen, J.; Ngo, J.; Hajkova, D.; Yeh, I.J.; Gama, V.; Miyagi, M.; Matsuyama, S. Cell-Penetrating Penta-Peptides (CPP5s): Measurement of Cell Entry and Protein-Transduction Activity. Pharmaceuticals 2010, 3, 3594–3613.
- Sheldrake, H.M.; Patterson, L.H. Function and Antagonism of β: Integrins in the Development of Cancer Therapy. Curr. Cancer Drug Targets 2009, 9, 519–540.
- Kadonosono, T.; Yamano, A.; Goto, T.; Tsubaki, T.; Niibori, M.; Kuchimaru, T.; Kizaka-Kondoh, S. Cell Penetrating Peptides Improve Tumor Delivery of Cargos through Neuropilin-1-Dependent Extravasation. J. Control. Release 2015, 201, 14–21.
- De, G.; Ko, J.K.; Tan, T.; Zhu, H.; Li, H.; Ma, J. Amphipathic Tail-Anchoring Peptide Is a Promising Therapeutic Agent for Prostate Cancer Treatment. Oncotarget 2014, 5, 7734–7747.
- Li, Z.; Cho, C. Development of Peptides as Potential Drugs for Cancer Therapy. Curr. Pharm. Des. 2010, 16, 1180–1189.
- Yin, H.; Zhang, Q.; Yang, J.; Wang, H.; Xu, J.; Zheng, J. IRGD as a Tumor-Penetrating Peptide for Cancer Therapy (Review). Mol. Med. Rep. 2017, 15, 2925–2930.
- Liu, S. Radiolabeled Multimeric Cyclic RGD Peptides as Integrin Αvβ3 Targeted Radiotracers for Tumor Imaging. Mol. Pharm. 2006, 3, 472–487.
- Danhier, F.; Le Breton, A.; Préat, V. RGD-Based Strategies to Target Alpha(v) Beta(3) Integrin in Cancer Therapy and Diagnosis. Mol. Pharm. 2012, 9, 2961–2973.
- Sugahara, K.N.; Teesalu, T.; Prakash Karmali, P.; Ramana Kotamraju, V.; Agemy, L.; Greenwald, D.R.; Ruoslahti, E. Coadministration of a Tumor-Penetrating Peptide Enhances the Efficacy of Cancer Drugs. Science 2010, 328, 1031–1035.
- Akashi, Y.; Oda, T.; Ohara, Y.; Miyamoto, R.; Kurokawa, T.; Hashimoto, S.; Enomoto, T.; Yamada, K.; Satake, M.; Ohkohchi, N. Anticancer Effects of Gemcitabine Are Enhanced by Co-Administered IRGD Peptide in Murine Pancreatic Cancer Models That Overexpressed Neuropilin-1. Br. J. Cancer 2014, 110, 1481–1487.
- Kulhari, H.; Pooja, D.; Kota, R.; Reddy, T.S.; Tabor, R.F.; Shukla, R.; Adams, D.J.; Sistla, R.; Bansal, V. Cyclic RGDfK Peptide Functionalized Polymeric Nanocarriers for Targeting Gemcitabine to Ovarian Cancer Cells. Mol. Pharm. 2016, 13, 1491–1500.
- Kuznetsov, A.V.; Margreiter, R.; Amberger, A.; Saks, V.; Grimm, M. Changes in Mitochondrial Redox State, Membrane Potential and Calcium Precede Mitochondrial Dysfunction in Doxorubicin-Induced Cell Death. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1144–1152.
- Pelicano, H.; Carney, D.; Huang, P. ROS Stress in Cancer Cells and Therapeutic Implications. Drug Resist. Update 2004, 7, 97–110.
- Alva, A.; Slovin, S.; Daignault, S.; Carducci, M.; DiPaola, R.; Pienta, K.; Agus, D.; Cooney, K.; Chen, A.; Smith, D.C.; et al. Phase II Study of Cilengitide (EMD 121974, NSC 707544) in Patients with Non-Metastatic Castration Resistant Prostate Cancer, NCI-6735. A Study by the DOD/PCF Prostate Cancer Clinical Trials Consortium. Investig. New Drugs 2012, 30, 749–757.
- Tang, Z.; Feng, W.; Yang, Y.; Wang, Q. Gemcitabine-Loaded RGD Modified Liposome for Ovarian Cancer: Preparation, Characterization and Pharmacodynamic Studies. Drug Des. Dev. Ther. 2019, 13, 3281–3290.
- Cai, W.; Geng, C.; Jiang, L.; Sun, J.; Chen, B.; Zhou, Y.; Yang, B.; Lu, H. Encapsulation of Gemcitabine in RGD-Modified Nanoliposomes Improves Breast Cancer Inhibitory Activity. Pharm. Dev. Technol. 2020, 25, 640–648.
- Yu, X.; Song, Y.; Di, Y.; He, H.; Fu, D.; Jin, C. Enhanced Tumor Targeting of CRGD Peptide-Conjugated Albumin Nanoparticles in the BxPC-3 Cell Line. Sci. Rep. 2016, 6, 1–9.
- Park, Y.H.; Jung, K.H.; Im, S.A.; Sohn, J.H.; Ro, J.; Ahn, J.H.; Kim, S.B.; Nam, B.H.; Oh, D.Y.; Han, S.W.; et al. Phase III, Multicenter, Randomized Trial of Maintenance Chemotherapy versus Observation in Patients with Metastatic Breast Cancer after Achieving Disease Control with Six Cycles of Gemcitabine plus Paclitaxel as First-Line Chemotherapy: KCSG-BR07-02. J. Clin. Oncol. 2013, 31, 1732–1739.
- Borsoi, C.; Leonard, F.; Lee, Y.; Zaid, M.; Elganainy, D.; Alexander, J.F.; Kai, M.; Liu, Y.T.; Kang, Y.; Liu, X.; et al. Gemcitabine Enhances the Transport of Nanovector-Albumin-Bound Paclitaxel in Gemcitabine-Resistant Pancreatic Ductal Adenocarcinoma. Cancer Lett. 2017, 403, 296–304.
- Yu, D.M.; Li, W.; Zhang, Y.; Zhang, B. Anti-Tumor Efficiency of Paclitaxel and DNA When Co-Delivered by PH Responsive Ligand Modified Nanocarriers for Breast Cancer Treatment. Biomed. Pharmacother. 2016, 83, 1428–1435.
- Noh, I.; Kim, H.O.; Choi, J.; Choi, Y.; Lee, D.K.; Huh, Y.M.; Haam, S. Co-Delivery of Paclitaxel and Gemcitabine via CD44-Targeting Nanocarriers as a Prodrug with Synergistic Antitumor Activity against Human Biliary Cancer. Biomaterials 2015, 53, 763–774.
- Zhang, J.; Zhang, P.; Zou, Q.; Li, X.; Fu, J.; Luo, Y.; Liang, X.; Jin, Y. Co-Delivery of Gemcitabine and Paclitaxel in CRGD-Modified Long Circulating Nanoparticles with Asymmetric Lipid Layers for Breast Cancer Treatment. Molecules 2018, 23, 2906.
- Han, H.; Jin, Q.; Wang, Y.; Chen, Y.; Ji, J. The Rational Design of a Gemcitabine Prodrug with AIE-Based Intracellular Light-up Characteristics for Selective Suppression of Pancreatic Cancer Cells. Chem. Commun. 2015, 51, 17435–17438.
- Bai, K.B.; Láng, O.; Orbán, E.; Szabó, R.; Köhidai, L.; Hudecz, F.; Mezö, G. Design, Synthesis, and in Vitro Activity of Novel Drug Delivery Systems Containing Tuftsin Derivatives and Methotrexate. Bioconjug. Chem. 2008, 19, 2260–2269.
- Liu, W.; Mao, Y.; Zhang, X.; Wang, Y.; Wu, J.; Zhao, S.; Peng, S.; Zhao, M. RGDV-Modified Gemcitabine: A Nano-Medicine Capable of Prolonging Half-Life, Overcoming Resistance and Eliminating Bone Marrow Toxicity of Gemcitabine. Int. J. Nanomed. 2019, 14, 7263–7279.


