 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Gabriele Centi | + 874 word(s) | 874 | 2021-01-14 09:47:51 | | | |
| 2 | Catherine Yang | Meta information modification | 874 | 2021-02-19 09:48:58 | | | | |
| 3 | Catherine Yang | -5 word(s) | 869 | 2021-02-19 09:50:24 | | |
Video Upload Options
Nitrogenase defines the familty of enzymes that catalyze the reduction of N2 (molecular nitrogen) to NH3 (ammonia). It is the only enzyme family which can realize this process of nitrogen fization.
1. Introduction
Nitrogenase is a unique system able to convert N2 to NH3 [1]. Total cost of N2 reduction corresponds to eight electrons being transferred and 16 MgATPs being hydrolyzed. Three classes of nitrogenase differ in the heteroatom present in the active site metal cluster (Mo, V or Fe), but the Mo-dependent nitrogenase is the most important and best-studied enzyme [2]. It contains two metallo-components, dinitrogenase (molybdenum–iron (MoFe) protein) and dinitrogenase reductase (Fe protein), which associate and dissociate in a catalytic cycle also requiring a reducing source and MgATP [3]. The MoFe protein contains two metal clusters: the iron–molybdenum cofactor (FeMo-co) [4], which provides the active site for substrate binding and reduction, and a P-cluster, involved in electron transfer from the Fe protein to FeMo-co [5]. The FeMo cofactor is thus the key element for the mechanism of N2 fixation. It also contains an interstitial carbon atom [6] which is key for the CO2 conversion mechanism, providing stability to the complex. Figure 1a reports the model of the FeMo cofactor, while Figure 1b shows the proposed mechanism of N2 fixation [1].
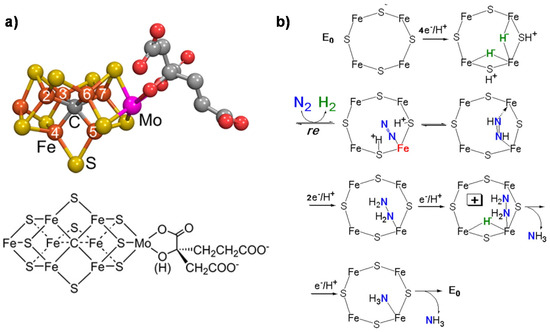
Figure 1. (a) FeMo cofactor (Fe in orange, Mo in magenta, S in yellow, C in gray and O in red); (b) mechanism (key elements) of N2 fixation on FeMo-co. Adapted from [1]. Not subject to US copyright.
The key features of this mechanism [7][8] are:
- A specific binding site for N2 able to first accept four electrons/protons to form two [Fe–H–Fe] bridging hydrides,
- coordination of N2 on two iron atoms with simultaneous reductive elimination of H2,
- multi-electron/proton transfer to a coordinated undissociated N2 molecule to form a N2H2 molecule stabilized by interaction with two iron atoms,
- further multi H+/e− transfer to form an end-on N2H4 coordinated molecule,
- further steps of H+/e− transfer with stepwise release of ammonia.
2. Mechanisms in Nitrogenase
The simultaneous electron/proton transfer to an undissociated coordinated N2 molecule is a different feature regarding the mechanisms in iron catalysts for the high-temperature/pressure HB (Haber-Bosch) process, where the first (rate-limiting) step is the dinitrogen dissociative chemisorption followed by sequential hydrogenation of the intermediates [9][10][11][12]. The reason for the difference in the mechanism is that the nitrogen molecule dissociation path, in principle, is preferable as sequential steps of electron transfer and hydrogenation of the nitrogen adatoms. However, the strongly bound nitrogen species formed by dissociation require high temperatures to avoid catalyst inhibition, and, therefore, there is a need for high-pressure operations (as this is an exothermic reversible reaction). For the undissociated N2 molecule activation, sequential electron and proton transfer steps would generate high-energy intermediates [13]. For example, adding one electron and one proton to N2 to form a N2H* species requires −3.2 V vs. normal hydrogen electrode (NHE). For multi-electron reduction processes, for example, to produce N2H5+ adspecies, the reduction potential becomes −0.23 V. Multi-electron reactions are thus necessary in the low-temperature profile, in agreement with what was observed for nitrogenase, and to avoid/minimize the side reaction of H+/e− recombination to form H2, which is also possible in the enzyme path.
As a slight change, more recent indications in the mechanism [14] instead suggest the formation of a Janus intermediate, i.e., the formation of a symmetric hydride structure with a subsequent diazene intermediate (Figure 2).
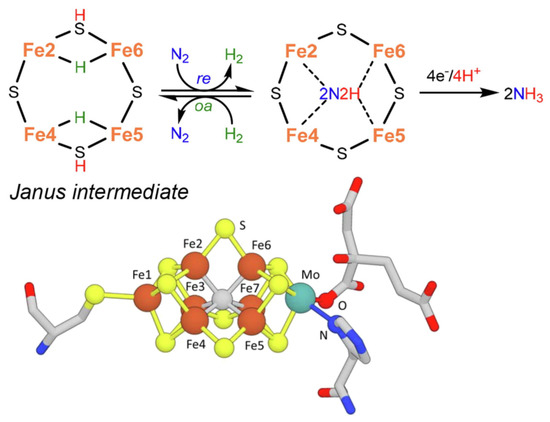
Figure 2. Key step for biological N2 fixation and structure of nitrogenase FeMo-co. Adapted from [14]. Copyright 2021 Elsevier.
Nitrogenase, particularly in the vanadium form [15], can also catalyze the reductive carbon–carbon coupling of COx into hydrocarbon products. This is also a unique feature and suggests a role of this enzyme as an evolutionary link between the nitrogen and carbon cycles on Earth [16]. The mechanism of C-C coupling is not clear, but progress has been made recently [17]. From CO2, the mechanism involves a first step of reduction of CO2 to CO by the Fe protein ([Fe4S4]0) of nitrogenase [18], followed by the reductive carbon–carbon coupling of two coordinated CO molecules on the same active site of the nitrogenase cofactor proposed for N2 fixation (M-cluster). The tentative mechanism is presented in Figure 3.
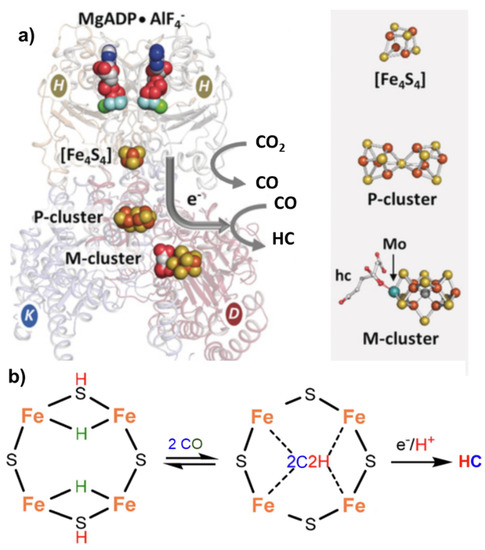
Figure 3. (a) Nitrogenase enzyme with indications of the key components involved in electron transfer, including MgADP AlF4−, [Fe4S4] cluster, P-cluster and M-cluster with indication of the active centers involved in CO2 to CO and CO2 to hydrocarbon (HC) conversion. Adapted from [16]. Copyright 2016 Wiley. (b) Proposed schematic mechanism of the active sites in FeMo cofactor (M-cluster) for conversion of CO to HC.
The same Janus intermediate proposed for N2 fixation (symmetric hydride structure) reacts with two CO molecules (produced on the Fe protein) to form an ethyne-like intermediate which can be hydrogenated to ethylene or could react further to form a ferracycle (formed by C2H4 binding to one of the Fe atoms [19]), leaving the other Fe free to form other Fe-hydride species and further coordinate CO, which can react with the ferracycle species, to give C2 products (hydrocarbon, oxygenated chemicals) (Figure 4).
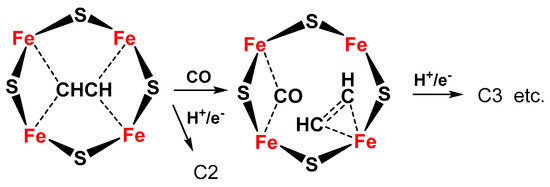
Figure 4. Proposed schematic mechanism of the active sites in FeMo cofactor for conversion of C2 intermediate to C3 products (hydrocarbon, oxygenates) via formation of a ferracycle species.
References
- Hoffman, B.M.; Lukoyanov, D.; Yang, Z.-Y.; Dean, D.R.; Seefeldt, L.C. Mechanism of Nitrogen Fixation by Nitrogenase: The Next Stage. Chem. Rev. 2014, 114, 4041–4062.
- Khare, E.; Yadav, A. The Role of Microbial Enzyme Systems in Plant Growth Promotion. Clim. Chang. Envir. Sustain. 2017, 5, 122–145.
- Burgess, B.K.; Lowe, D.J. Mechanism of Molybdenum Nitrogenase. Chem. Rev. 1996, 96, 2983–3011.
- Burgess, B.K. The iron-molybdenum cofactor of nitrogenase. Chem. Rev. 1990, 90, 1377–1406.
- Kim, J.; Rees, D.C. Structural models for the metal centers in the nitrogenase molybdenum-iron protein. Science 1992, 257, 1677–1682.
- Spatzal, T.; Aksoyoglu, M.; Zhang, L.; Andrade, S.L.A.; Schleicher, E.; Weber, S.; Rees, D.C.; Einsle, O. Evidence for Interstitial Carbon in Nitrogenase FeMo Cofactor. Science 2011, 334, 940.
- Khadka, N.; Milton, R.D.; Shaw, S.; Lukoyanov, D.; Dean, D.R.; Minteer, S.D.; Raugei, S.; Hoffman, B.M.; Seefeldt, L.C. Mechanism of Nitrogenase H2 Formation by Metal-Hydride Protonation Probed by Mediated Electrocatalysis and H/D Isotope Effects. JACS 2017, 139, 13518–13524.
- Lukoyanov, D.; Yang, Z.-Y.; Khadka, N.; Dean, D.R.; Seefeldt, L.C.; Hoffman, B.M. Identification of a Key Catalytic Intermediate Demonstrates That Nitrogenase Is Activated by the Reversible Exchange of N2 for H2. JACS 2015, 137, 3610–3615.
- Garden, A.L.; Skúlason, E. The Mechanism of Industrial Ammonia Synthesis Revisited: Calculations of the Role of the Associative Mechanism. J. Phys. Chem. C 2015, 119, 26554–26559.
- Fuller, J.; Fortunelli, A.; Goddard III, W.A.; An, Q. Reaction mechanism and kinetics for ammonia synthesis on the Fe(211) reconstructed surface. Phys. Chem. Chem. Phys. 2019, 21, 11444–11454.
- Schlögl, R. Catalytic synthesis of ammonia-a “never-ending story”? Angew. Chem. Int. Ed. 2003, 42, 2004–2008.
- Schütze, J.; Mahdi, W.; Herzog, B.; Schlögl, R. On the structure of the activated iron catalyst for ammonia synthesis. Topics Catal. 1994, 1, 195–214.
- Bazhenova, T.; Shilov, A. Nitrogen fixation in solution. Coord. Chem. Rev. 1995, 144, 69–145.
- Ghosh, A.C.; Duboc, C.; Gennari, M. Synergy between metals for small molecule activation: Enzymes and bio-inspired complexes. Coord. Chem. Rev. 2021, 428, 213606.
- Rebelein, J.G.; Hu, Y.; Ribbe, M.W. Differential Reduction of CO2 by Molybdenum and Vanadium Nitrogenases. Angew. Chem. Int. Ed. 2014, 53, 11543–11546.
- Hu, Y.; Ribbe, M.W. Nitrogenases—A Tale of Carbon Atom(s). Angew. Chemie Int. Ed. 2016, 55, 8216–8226.
- Seefeldt, L.C.; Yang, Z.-Y.; Lukoyanov, D.A.; Harris, D.F.; Dean, D.R.; Raugei, S.; Hoffman, B.M. Reduction of Substrates by Nitrogenases. Chem. Rev. 2020, 120, 5082–5106.
- Rettberg, L.A.; Stiebritz, M.T.; Kang, W.; Lee, C.C.; Ribbe, M.W.; Hu, Y. Structural and Mechanistic Insights into CO2 Activation by Nitrogenase Iron Protein. Chem. A Europ. J. 2019, 25, 13078–13082.
- Lee, H.-I.; Sorlie, M.; Christiansen, J.; Yang, T.-C.; Shao, J.; Dean, D.R.; Hales, B.J.; Hoffman, B.M. Electron Inventory, Kinetic Assignment (En), Structure, and Bonding of Nitrogenase Turnover Intermediates with C2H2 and CO. JACS 2005, 127, 15880–15890.


