1000/1000
Hot
Most Recent
 +1 point
+1 point
Calcium (Ca2+) is a major second messenger in cells and is essential for the fate and survival of all higher organisms. Different Ca2+ channels, pumps, or exchangers regulate variations in the duration and levels of intracellular Ca2+, which may be transient or sustained. These changes are then decoded by an elaborate toolkit of Ca2+-sensors, which translate Ca2+ signal to intracellular operational cell machinery, thereby regulating numerous Ca2+-dependent physiological processes. Alterations to Ca2+ homoeostasis and signaling are often deleterious and are associated with certain pathological states, including cancer. Altered Ca2+ transmission has been implicated in a variety of processes fundamental for the uncontrolled proliferation and invasiveness of tumor cells and other processes important for cancer progression, such as the development of resistance to cancer therapies.
In resting cells, the intracellular free Ca2+ concentration ([Ca2+]i) is maintained at lower levels than extracellular fluid. Indeed, there is a 20,000-fold gradient between outside (about 1.2 mM) and inside (approximately 10–100 nM) of cells. Moreover, in the mitochondria and in the nucleus, the concentrations of Ca2+ are similar to those in the cytoplasm. In the endoplasmic reticulum (ER), considered the main intracellular Ca2+ store, the [Ca2+] ranges between 100 and 800 μM [1]. In addition, direct measurements of Ca2+ levels show that lysosomes present an internal [Ca2+] of about ≈500 μM [2]. Therefore, it exists an elaborate system of Ca2+-transporters, -channels, -exchangers, -binding/buffering proteins, and -pumps that finely regulate Ca2+ flow inside and outside of cells and among intracellular organelles [3]. This network permits preservation of a low resting [Ca2+] and regulates the propagation of intracellular Ca2+ changes that are fundamental to intracellularly transmitted biological information and important physiologic processes, including metabolism, cell proliferation and death, protein phosphorylation, gene transcription, neurotransmission, contraction, and secretion [4][5]. During cell stimulation the [Ca2+]i can increase more than twofold at the micromolar level. Different channels situated in the plasma membrane (PM) induce the influx of extracellular Ca2+ into the cells. Among these channels, the most important are transient receptor potential channels (TRPC) [6], store-operated Ca2+ entry (SOCE) channels such as ORAI and STIM [7], voltage-gated Ca2+ channels (VGCC) in excitable cells [8], receptor-operated Ca2+ channels such as the N-methyl-d-aspartate receptor (NMDA) [9] and purinergic P2 receptors [10], whose activation determines cytosolic Ca2+ influx. Intracellular Ca2+ increases may be also due to Ca2+ release from internal stores, mainly via inositol 1,4,5-triphosphate receptors (IP3Rs) situated on the ER [11][12]. IP3Rs are large-conductance cation channels that are activated in response to the activation of cell surface receptors [13]. Despite different physiological and pharmacological profiles, ryanodine receptors (RyRs) have an approximatively 40% homology with IP3Rs and are the Ca2+ release channels on the sarcoplasmic reticulum of muscle cells [14]. A prolonged elevation of [Ca2+]i has adverse effects for the cells. Therefore, different channels, pumps, and buffering systems reestablish low [Ca2+]i. The reuptake of Ca2+ into the ER lumen is allowed by the activity of sarcoendoplasmic reticulum Ca2+-ATPase (SERCA), which pumps Ca2+ into the ER with a stoichiometry of 2:1 Ca2+/ATP and by the secretory protein calcium ATPase (SPCA), which transports Ca2+into the Golgi apparatus [15]. Plasma membrane Ca2+ transport ATPase (PMCA) and Na+/Ca2+ exchanger (NCX) are the two mechanisms situated on the PM responsible for Ca2+ extrusion. PMCA is a pump that belongs to the class of P-type ATPases that pump Ca2+ across the PM out of the cell at the expense of ATP [16][17]. NCX permits Ca2+ extrusion against its gradient without energy consumption by using the electrochemical gradient of Na+. For each Ca2+ ion extruded, three Na+ ions enter the cell [18]. Additionally, mitochondria significantly contribute to the signaling pattern of released intracellular Ca2+. Indeed, these organelles may act as Ca2+ buffers [19]. It is widely accepted that Ca2+ entry into mitochondria is mediated by the activity of the mitochondrial calcium uniporter (MCU) complex, composed of the pore-forming subunit of the MCU channel together with several regulatory proteins (MICU1, MICU2, MICU3, MCUR1, MCUb, and EMRE) [20]. Advances in the studies regarding Ca2+ dynamics have revealed that a network of membrane contact sites has a determinant role in Ca2+ signaling. These contacts create microdomains that permit the exchange of metabolites and signals between membranes of different compartments. The structural and functional interactions between the ER and mitochondria (the mitochondria associated membranes, MAMs) represent the main central hub for controlling Ca2+ exchange between these two compartments [21]. Disruption of MAMs result in the suppression of ER Ca2+-release and alters mitochondrial Ca2+ accumulation (Figure 1). ER membranes are also interconnected with the membranes of lysosomes to form the ER-lysosome membrane contact sites. It has been proposed that the IP3R-mediated ER release of Ca2+ is a mechanism for mediating the reestablishment of Ca2+ levels in lysosomes [22]. However, the Ca2+ transporter mediating this Ca2+ transmission remains unidentified. In contrast, the identity of channels regulating lysosomal Ca2+ release has been established. Several channels mediate this Ca2+ transport. Among these channels, the mucolipin subgroup of the TRP ion channel family, in particular the isoform TRPML1, represents the most well-established lysosomal Ca2+ release channels [23].
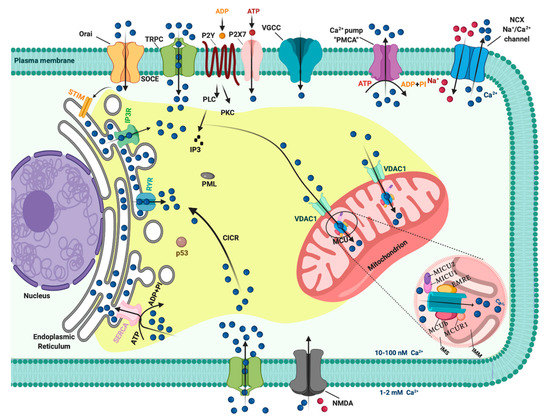
Figure 1. The intracellular Calcium (Ca2+) signaling. Different Ca2+ transporters, channels, exchangers, binding/buffering proteins and pumps mediate the regulation of cytosolic Ca2+ concentration. In the plasma membrane (PM), PM Ca2+-ATPases (PMCA) pumps, transient receptor potential channels (TRPC), voltage-gated Ca2+ channels (VGCC), Na+/Ca2+ exchanger (NCX), and purinergic P2 receptors regulate the transport of Ca2+ ions inside and outside cells. Inositol 1,4,5-triphosphate receptors (IP3R), ryanodine receptors (RyR), and sarcoendoplasmic reticulum Ca2+-ATPase (SERCA) pumps control the storage of Ca2+ in the endoplasmic reticulum. Finally, voltage-dependent anion channels (VDAC) and members of the mitochondrial Ca2+ uniporter family are critical for controlling the mitochondrial Ca2+ uptake. Created with BioRender.com.
In recent years, the importance of cell cycle progression regulating by Ca2+ signals has been recognized, especially upon the development of probes that allow a very sensible and direct visualization of Ca2+ transients. Spontaneous Ca2+ oscillations at the three major cell cycle checkpoints have been described. For example, transient [Ca2+]i increases during the G1/S phase transition [24], the G2/M transition [25], and the metaphase to anaphase transition [26]. Ca2+ is also required in the early G1 phase, when cells re-enter the cell cycle, to promote the activation of c-AMP-responsive element binding protein, AP1 (FOS and JUN) transcription factors, and the nuclear factor of activated T-cell (NFAT) [27]. The cell cycle is principally controlled by the expression of protein complexes organized around cyclin-dependent protein kinases (CDKs), which coordinate the entry into the next phase of the cell cycle only when bound to a cyclin. The essential bridge between Ca2+ ions and CDK/cycline complexes is undoubtedly represented by the Ca2+-sensors calmodulin (CAM) and calcineurin (CaN). These Ca2+-binding proteins, and intermediary proteins such as Ca2+/calmodulin-dependent protein kinases (CAMKI, CAMKII, and CAMKIII), interact with CDK/cycline complexes regulating crucial cell cycle events, including DNA synthesis (i.e., by cyclin D1-CDK4 regulation through CAMKI) [28], microtubule stability regulation, e.g., by decreasing the amount of Ca2+ required for microtubule depolymerization [29] and by interacting with nucleoporin p62 [30] and for cytokinesis completion [31]. The essential role played by Ca2+ as a regulator of cell cycle progression has been extensively presented in publications based on the use of CAM activity inhibitors. In particular, treatment with W-7 and W-13 CAM antagonists induced G1 phase cell cycle arrest by downregulating cyclins and upregulating p21 [32]. Moreover, the microinjection of monoclonal antibodies against CAM inhibited the synthesis of DNA in a dose-dependent manner [33]. One of the most fascinating aspects of studying these cell cycle progression regulation mechanisms is undoubtedly the detection of new potential targets in the fight against cancer. Indeed, the most important characteristic of cancer cells is certainly their ability to undergo biological changes that sustain their unlimited replicative capacities. The behavioral study of Ca2+ channels and pumps in relation to the cell cycle and proliferation has turned out to be very important, especially in recent years. Some examples of how a perturbation of Ca2+ signaling can lead to cell cycle dysregulation with consequent repercussions on tumor pathologies are worthy of description. Cytosolic Ca2+ levels modulate guanosine exchange factor and GTPase activating protein, which are a RAS stimulator and a RAS inhibitor, respectively. RAS, in turn, stimulates the proliferative mitogen-activated protein kinase (MAPK) pathway, which initiates the cells transition into the S phase because of phosphorylation of the tumor suppressor RB1 upon cytoplasmic cyclin D1 upregulation. Constitutively high cytosolic Ca2+ levels in cancer cells can lead to uncontrolled growth through the removal of the G1/S transition checkpoint [34]. ORAI3, a SOCE component, is overexpressed in breast cancer biopsy samples and is involved in breast cancer cell proliferation and cell cycle progression by modulating the G1 phase and G1/S transition regulator protein activity [35]. ORAI3 is an upstream regulator of c-myc that controls the cell cycle and proliferation in breast cancer by modulating the expression of cyclins D1 and E, CDKs 4 and 2, cyclin-dependent kinase inhibitor p21, and tumor-suppressing protein p53 [36]. A large number of studies have indicated key roles for cyclin D and cyclin E expression in breast cancer cell cycle deregulation; in fact, cyclins D1 and E proteins are overexpressed in more than 50% of breast tumors [37][38][39]. Additionally, VGCCs are associated with cell proliferation regulation. Specific VGCCs family genes were downregulated in breast, kidney, brain, and lung cancers, showing that these Ca2+ channels play roles as tumor suppressor genes [40]. On the other hand, members of the VGCCs family are expressed at detectable levels in melanoma cells but not in untransformed melanocytes, and the use of T-type channel inhibitors induces cell cycle arrest with a significant increase of the percentage of cells in the G1 phase and a reduction of cells in the S phase [41].
Changes in the expression of TRPCs have been implicated in prostate cancer. In particular, transient receptor potential vanilloid subfamily member 6 (TRPV6) in prostate cancer reduces the activation of NFAT and decreases cell accumulation in the S phase of the cell cycle [42].
It has been reported that CAMKs expression alterations have repercussions on cell cycle progression in several tumor pathologies. Parmer et al. described CAMKIII as a potential pharmacological target against glioma because of its important link to cell proliferation, viability, and malignancy [43]. Chemical inhibition of CAMKIII resulted in the reduction of growth of glioma cells line, which was mirrored by a blocked G1 phase transition in the cell cycle. In breast cancer, CAMKIII activation leads to the phosphorylation of elongation factor-2 and transient inhibition of protein synthesis. These events are controlled by mitogens and are predominant in the S phase of the cell cycle [44]. CAMKII has been described to be crucial in T cell lymphoma cell proliferation; its genetic ablation drives a significant increase in the percentage of G2/M phase cells and a decrease in the percentage of S phase cells, outcomes that are consistent with the inhibition of cell proliferation [45].
A main hallmark of cancer cells is evasion of programmed cell death (PCD) [46]. PCD is a genetically determined cell routine in which cells undergo an unexpected decline in homeostasis and functionality, triggering several intracellular pathways and ultimately cell death. Different types of PCD exists. During viral or microbial infections, PCD is a part of the host immune response, with traits similar to those of apoptosis and necrosis in a process referred to as pyroptosis or necroptosis. Pyroptosis is a caspase-dependent form of PCD that leads to membrane permeabilization and cell swelling through gasdermin D activation. Pyroptosis is triggered via inflammasome that was induced when the cell senses changes after viral or microbial invasion and is linked to atherosclerosis, metabolic disease, and neuroinflammatory disorders [47][48]. Another type of PCD is necroptosis, an inflammation-dependent form of PCD, which is similar to necrosis in the absence of caspase involvement and eventual membrane permeabilization, cell swelling, and lysis, with the subsequent leakage of a plethora of proinflammatory molecules. [49]. However, the best-characterized form of PCD is apoptosis. Apoptosis is a strictly controlled phenomenon, typically manifested by chromatin condensation, DNA and nuclear fragmentation, mitochondrial failure, proteolytic enzyme activation and membrane blebbing, even with cell membrane integrity. The cell membrane forms the interface with cell-impermeant stimuli, which interact with membrane receptors triggering the so-called intrinsic pathways. Thus, membrane-induced apoptosis depends upon extracellular ligands, such as tumor necrosis factor-α (TNFα) and first apoptosis signal (FAS) ligand, and their receptors, TNFRs and FAS. Employing several adaptors, some of which carry the death effector domain, these activated receptors induce the formation of the death-inducing signaling complex (DISC). DISC stimulates the autoproteolytic cleavage of initiator caspase-8, which in turn activates the executioner caspase-3, -6, and -7. This proteolytic cascade strongly amplifies the initial signal and initiates the cleavage of hundreds of cellular targets and is thus crucial for the main morphological features of apoptosis [50] (Figure 2, upper panel).
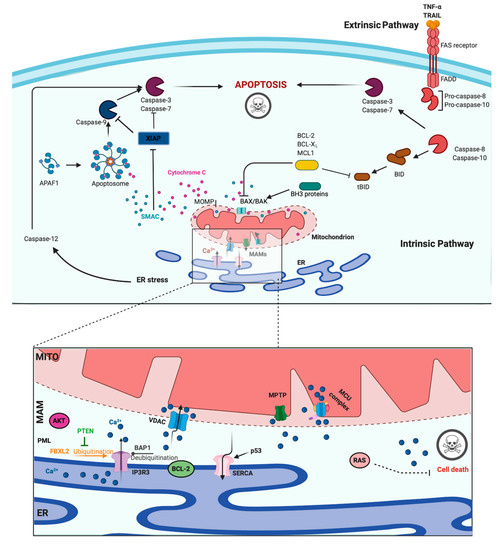
Figure 2. Apoptosis and Calcium (Ca2+) dynamics in cancer. Apoptosis is the best-characterized and studied programmed cell death. In the extrinsic pathway, extracellular ligands determine the formation of the death-inducing signaling complex that activates the caspases cascade. The intrinsic apoptotic pathway is characterized by permeabilization of the mitochondria that allows the release of cytochrome c (cyt-c) and other apoptogenic factors in the cytosol. Once released, these factors bind apoptotic protease activating factor 1 (APAF1) and form a multiprotein complex called the apoptsome that recruits and activates the caspases. Ca2+ has a major role during intrinsic apoptosis, and excessive mitochondrial Ca2+ accumulation may trigger apoptosis. Different proteins were found to control apoptotic machinery by regulating Ca2+ flux between endoplasmic reticulum (ER) and mitochondria. Antiapoptotic B-cell lymphoma-2 (BCL-2) members block apoptotic program by lowering Ca2+ levels in the ER, thereby attenuating subsequent Ca2+ release. p53 localizes at the ER–mitochondria interface to improve Ca2+ dynamics and apoptosis by increasing sarcoendoplasmic reticulum Ca2+-ATPase (SERCA) pumps activities. Additionally, the tumor suppressors promyelocytic leukemia protein (PML), BRCA1-associated protein 1 (BAP1), and phosphatase and tensin homolog (PTEN) move to the mitochondria associated membranes (MAMs) to regulate Ca2+-dependent apoptosis. They determine the activation of Ca2+ release from the ER by modulating the activity of inositol 1,4,5-triphosphate receptor 3 (IP3R3). Mutations or loss of these tumor suppressors are frequently found in diverse human tumor samples, where they lead to a reduction in Ca2+ homeostasis and the apoptosis rate, favoring cellular proliferation, tumor growth, maintenance, and metastasis. Created with BioRender.com.
In addition to the extracellular-driven intrinsic pathway, an intracellular, mitochondria-centered pathway is activated in apoptosis, with the function of coupling insurmountable mitochondrial stress to cell death. Nevertheless, a wide number of stress conditions, including hypoxia, alteration, or poisoning of the electron transfer chain, unbuffered ROS production, and imbalanced mitochondrial protein homeostasis, can initiate mitochondrial permeability transition (MPT) [51][52]. MPT, followed by mitochondrial osmotic imbalance and mitochondrial outer membrane permeabilization, allows the release of several mitochondrial proteins, such as cytochrome C (cyt-C). In particular, cytoplasmic cyt-C binds to apoptotic protease activating factor 1 to form a multiprotein complex able to recruit and activate the initiator caspase-9 via the caspase recruitment domain. Caspase-9 in turn cleaves and activates the other executioners, namely, caspase-3, -6, and -7 [50]. An important regulatory mechanism of the intrinsic pathway is the mitochondrial Ca2+ load. Ca2+ is an important regulator of Krebs cycle dehydrogenases [53] and normally accumulated in the mitochondrial matrix at concentrations 10-fold higher than those measured in the cytosol. Under specific conditions, Ca2+ overload can trigger MTP by opening the permeability transition pore with the consequent release of apoptogenic factors [51][54] (Figure 2, upper panel). During carcinogenesis, cancer cells use different machinery to circumvent apoptosis and acquire a profound survival and proliferative advantage. They accumulate genetic alterations that increase or decrease the expression of pro- and/or antiapoptotic genes. Moreover, cancer cells can prevent apoptosis through post-translation modification, such as phosphorylation/dephosphorylation. In contrast, cancer cells may also evade apoptosis by reducing the Ca2+ signaling necessary to prompt the apoptotic machinery. The latest evidence shows that Ca2+ release from the ER is the main mechanism regulating the mitochondrial Ca2+ remodeling and apoptosis [55]. The first observation was obtained by studying B-cell lymphoma-2 (BCL-2) proteins. These proteins are classified into antiapoptotic category (BCL-2, BCL-xL, and Mcl-1) and a pro-apoptotic category (like Bax, Bak, Bim, Bid, etc.). Evidence demonstrates that antiapoptotic BCL-2 proteins regulate the apoptotic program by controlling ER-mitochondrial Ca2+ transfer in both organelles, and in particular, recent studies indicate that these proteins also exert antiapoptotic functions at MAMs levels [56]. Overexpression of pro-apoptotic BCL-2 proteins was found to reduce both ER-Ca2+ release either by direct control of IP3R3-induced pore opening or by lowering the Ca2+ content of the ER [57][58]. As a consequence, Ca2+-induced MPT is prevented, and the apoptotic program is abolished. Furthermore, it has also been demonstrated that BCL-2, BCL-XL, and Mcl-1 determine pro-survival IP3Rs-mediated Ca2+ oscillations that are necessary to increase mitochondrial energy production and stimulate cell proliferation [59]. Overall, these proteins impact three important aspects of cancer development: cell death, survival, and energy production. Consistently, upregulation of pro-apoptotic BCL-2 members was found in different human cancer samples and was associated with the invasion and metastasis of colon, breast, and gastric cancer [60]. BCL-2 members are not the only proteins that regulate apoptosis and cell proliferation by modulating the ER-Ca2+ release into mitochondria. The oncogene RAS plays a pivotal role in tumor growth and maintenance of the tumor environment [61]. To exert this function, RAS deregulates ER Ca2+ dynamics with the consequent inhibition of apoptosis, impairment to mitochondrial metabolism, and promotion of malignant cell survival [62]. Additionally, the oncogene AKT phosphorylates and inactivates several proteins (such as Bad, Bax, and hexokinase-2) that normally work to promote the Ca2+-dependent apoptotic response. Furthermore, AKT inhibits the apoptotic process by exerting a direct control of IP3R3 opening, thus avoiding the Ca2+ overload necessary to activate the intrinsic apoptosis [63]. If oncogene proteins promote cell survival and proliferation by blocking Ca2+-mediated apoptosis, it is not surprising that tumor suppressors activate the same mechanism. Protein phosphatase and tensin homolog (PTEN), which is frequently lost or mutated in several cancers, counteracts the activity of AKT and restores Ca2+ transfer and reestablishes subsequent cell death [64]. In addition, PTEN was also recently found to block the proteosomal degradation of IP3R3 provoked by the F-box protein FBXL2 [65]. The activity of AKT is also balanced by the tumor suppressor promyelocytic leukemia protein (PML), which, together with IP3R3, AKT, and the phosphatase PP2a, creates a complex that rules ER-mitochondria Ca2+ transfer [66]. BRCA1-associated protein 1 (BAP1) is a tumor suppressor frequently mutated in diverse malignancies, especially in mesotheliomas, for which alteration of Ca2+ dynamics had previously described [67]. BAP1 works as a deubiquitinating enzyme and is involved in different processes, such as DNA repair and transcription. Recently, it has been demonstrated that BAP1 also deubiquitylates and stabilizes IP3R3. Therefore, following DNA damage exposure, cells can undergo to apoptosis by activating ER-mitochondria Ca2+ transfer [68]. Additionally, p53 regulates tumorigenesis by modulating ER-mitochondria Ca2+ flux. In this case, the tumor suppressor was found to improve intracellular Ca2+ accumulation by increasing SERCA pump activities [69] (Figure 2, lower panel). Apart from the well-established roles for ER-Ca2+ dynamics in cancer, recent investigations suggest that impairments in lysosomal Ca2+ processes are also important in driving tumorigenesis. Consistent with this finding, cancers of the bladder, head and neck region, and thyroid exhibit increased expression of the gene mucolipin 1 [70], which encodes the lysosomal Ca2+ release channel (transient receptor potential mucolipin 1, TRPML1). Consistent with this, TRPML1 inhibition reduces the proliferation of cancer cells [70]. Additionally, the expression of TRPML2 isoforms has been found to be highly expressed in glioma tissues [71]. The transcription factor EB (TFEB) is a master regulator of lysosome function. Altered expression and/or activity of TFEB has been found in pancreatic, kidney, and non-small cell lung cancers [72][73][74] and is associated with aggressive clinical features in colorectal cancer [75]. Interestingly, it has been demonstrated that TFEB activities are highly modulated by a Ca2+-enriched microenvironment that is created following lysosomal Ca2+ release mediated by TRPML1 channels [76] and that TFEB itself modulates the lysosomal Ca2+ buffering capacity [77], thereby suggesting a primary role of lysosomal Ca2+ in TFEB-associated cancers. Recent advances in RNA research have revealed that the levels of microRNAs (miRs), a class of small noncoding RNAs that regulate various target genes leading to a decrease in target protein levels, are associated with a variety of human diseases, including cancer. In this context, miRs not only regulate the functions of several oncogenes and tumor suppressors but also target genes that control intracellular Ca2+ dynamics. Among these miRNAs, oncogenic miR-25 provokes the downregulation of MCU with subsequent decreases in mitochondrial Ca2+ uptake and a reduction in the apoptotic process. Accordingly, prostate and colon cancer cells express increased miR-25 levels and present reduced MCU levels [78]. miR-25-dependent MCU downregulation has also been observed in pulmonary artery smooth muscle cells, where decreases in mitochondrial Ca2+ levels cause the activation of a cancer-like phenotype characterized by increased cellular proliferation, migration, and apoptotic resistance. In addition to miR-25, the authors also identified miR-138 as a regulator of MCU expression and demonstrated that nebulizing anti-miR-25 and miR-138 restored MCU expression and abolished the cancer-like phenotype [79]. Another miR involved in cancer is miR-34, whose aberrant expression has been detected in the T lymphocytes of cancer patients [80][81]. It has been observed that miR-34 is also a regulator of SOCE by targeting the expression of IP3R2, STIM1, and ORAI3 in immune cells. These results suggest that miR-34 may control the activities of pro- and antiapoptotic genes by regulating Ca2+ signaling, thereby controlling the activation and proliferation of T cells and inducing the inhibition of the antitumor immune response. Finally, several other miRs have been suggested to regulate Ca2+ homeostasis and apoptosis. Despite this, a direct correlation between these miRs, Ca2+, and cancers has not been demonstrated. Only to cite a few, miR-132 influences Ca2+ levels by regulating the expression of the exchanger NCX [82]. MiR-7 reduces voltage-dependent anion channels 1 (VDAC1) expression and diminishes the efflux of Ca2+ from mitochondria [83]. MiR-1 regulates the expression of MCU and protects mitochondria from Ca2+ overload in cardiac myocytes during development [84].


