 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mounia Chami | + 4588 word(s) | 4588 | 2021-01-26 04:55:05 | | | |
| 2 | Peter Tang | -2 word(s) | 4586 | 2021-02-06 09:49:53 | | |
Video Upload Options
The alteration of the physical association between the endoplasmic reticulum (ER) and mitochondria, also referred as mitochondria-associated membranes (MAMs), impacts various cellular housekeeping functions such as phospholipids-, glucose-, cholesterol-, and fatty-acid-metabolism, as well as calcium signaling, which are all altered in Alzheimer's disease (AD).
1. Introduction
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by two major histological hallmarks: (1) the neurofibrillary tangles (NFTs) corresponding to intracellular aggregates of abnormally hyperphosphorylated Tau protein (pTau) and, (2) senile plaques that are mainly composed of extracellular aggregates of β amyloid peptide (Aβ) derived from the sequential cleavage of its precursor, the amyloid precursor protein (APP), by β-secretase and γ-secretase enzymes [1][2]. Late-onset AD (LOAD) cases are considered to be sporadic (SAD) accounting for the majority of AD cases. For a long time, apolipoprotein E (APOE) has been described as the sole and main genetic risk factor for LOAD forms [3]. Recently, a genome wide-association study (GWAS) identified several new AD genetic risk factors [4][5][6]. Dominantly inherited familial AD (FAD) accounts for less than 1% of the cases and can be caused by mutations in APP, presenilin 1 (PS1), or presenilin 2 (PS2) genes [7][8]. Overall, AD is now considered a complex pathology, the mechanistic defects of which remain unclear. The amyloid cascade is the most widely accepted hypothesis in AD and proposes Aβ accumulation as the etiological trigger of the pathology. This has been supported by genetic evidence demonstrating that FAD mutations in genes coding for APP or its processing enzymes (PS1 and PS2) constituting the catalytic components of the γ-secretase enzymatic complex [9] lead to increased production and secretion of Aβ peptides [9]. The latter accumulate in the form of neurotoxic Aβ oligomers (Aβo) and are thought to trigger several stress responses in neurons leading to the onset of neurofibrillary degeneration [9]. Several therapeutic approaches were developed with the aim to reduce the accumulation of Aβ through active and passive immunizations. However, the failure of these trials to rescue cognitive declines or even stabilize them [10] casts some doubts about the amyloid cascade hypothesis. Notably, the sole contribution of Aβ to AD pathogenesis is challenged, since APP processing yields several fragments besides Aβ [2]. In physiological conditions, 90% of mature APP is cleaved by α-secretase at the plasma membrane, producing the secreted soluble α-APP (sAPPα) and the membrane-anchored APP-C-terminal (APP-CTF)α fragment (C83). The latter is further cleaved by γ-secretase producing p3 and AICD (APP intracellular domain) peptides. On the other hand, 10% of mature APP is cleaved by β-secretase (BACE1) following its internalization towards the endosomal/lysosomal pathway, producing the soluble β-APP (sAPPβ) released in the extracellular environment, and the membrane-anchored APP-CTFβ fragment (C99), which is further cleaved by γ-secretase producing AICD and Aβ peptides [2]. C99 is also cleaved by α-secretase to produce C83 [11]. Furthermore, other APP-derived fragments produced by other enzymes (such as η- and δ- secretases) were recently described [12][13][14], likely contributing to AD pathophysiology [12][13][14][15][16][17][18].
Both the endoplasmic reticulum (ER) and mitochondria are vital organelles of the cell. Rough and smooth ER participate in protein synthesis, folding, and transport. The ER is also involved in other fundamental cellular functions such as lipids- and carbohydrates- metabolism and is the major calcium (Ca²+) storage pool [19]. On the other hand, mitochondria act as the powerhouse of the cell by generating energy through adenosine triphosphate (ATP) production [20]. Besides, mitochondria play a major role in buffering the Ca²+ flux from the ER, but also in lipid- and amino acid- metabolism, beneficial and harmful reactive oxygen species (ROS) production, and apoptosis [20]. It is now well established that ER and mitochondria functions are highly interconnected. They physically interact to form specific microdomains called mitochondria-associated ER membranes (MAMs), where the outer mitochondrial membrane (OMM) is close to the ER in the order of 10–100 nanometers [21][22][23]. The maintenance of stable contact sites between the ER and mitochondria provides a platform for bidirectional crosstalk. Not surprisingly, MAMs control intracellular elementary events such as Ca²+ homeostasis, metabolic flow, protein transport [24], mitochondrial lipids production, phospholipid biosynthesis, mitochondrial fusion and fission, and global events such as autophagy and apoptosis [25] (Figure 1). In addition, MAMs are a hot-spot for the transfer of stress signals from the ER to mitochondria, particularly under ER stress conditions activating the unfolded protein response (UPR) [26] (Figure 1). As a result, one can argue that the alteration of ER and mitochondria communication may influence and disrupt these functions, thus leading to the development of several pathologies and vice-versa.
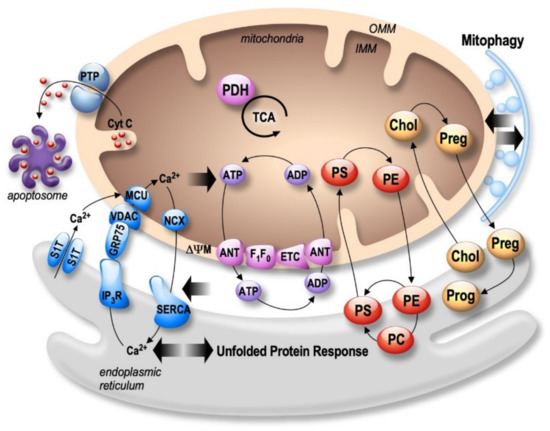
Figure 1. Bidirectional regulation of ER and mitochondria functions in MAMs. Mitochondria Ca2+ uptake (i.e., by the VDAC (voltage-dependent anion channel) located in the outer mitochondria membrane (OMM) and the mitochondrial Ca2+ uniporter complex (MCU) located in the inner mitochondria membrane (IMM)) increases the activity of mitochondrial enzymes (i.e., the pyruvate dehydrogenase (PDH), and the tricarboxylic acid cycle (TCA)) orchestrating the activity of the oxidative phosphorylation electron transport chain (ETC) producing ATP by the F1F0 ATP-synthase. ATP is then transported through the adenine nucleotide translocator (ANT). In return, ATP is used by the sarco-endoplasmic reticulum Ca2+ ATPase (SERCA) ensuring active Ca2+ storage in the ER, necessary for ER functions (i.e., protein synthesis and maturation). Mitochondrial Ca2+ homeostasis results from an equilibrium between Ca2+ uptake and extrusion by the Na2+/Ca2+ exchanger (NCX). Prolonged or excessive mitochondrial Ca²+ uptake triggers mitochondrial membrane permeability transition pore (PTP) opening, initiating cytochrome c (Cyt c) release, apoptosome formation, and the activation of apoptotic cell death. In particular, altered ER Ca2+ homeostasis is associated with the unfolded protein response (UPR), triggering excessive transfer of Ca2+ from the ER to mitochondria (i.e., through SERCA1 truncated isoform (S1T), or the inositol 1,4,5-trisphosphate receptors (IP3R)), which is tightly linked to ER stress-mediated mitochondria apoptotic cell death. In parallel, MAMs control phospholipids biosynthesis and transfer between organelles where phosphatidylcholine (PC); phosphatidylethanolamine (PE); and phosphatidylserine (PS) are the most abundant ones. MAMs are also the intersection site of transport and metabolism of cholesterol (Chol), which is catalyzed to generate pregnenolone (Preg) and released to the ER to produce progesterone (Prog). MAMs-associated lipid microdomains regulate the autophagic/mitophagic processes through the recruitment of autophagic proteins and autophagosome formation.
ER stress and mitochondrial dysfunctions occur early in AD, likely contributing to disease progression and irreversible neuronal death [27][28][29][30]. Numerous papers reported that several pathogenic paradigms associated with AD are closely linked to MAMs structure and function alterations.
2. Structural and Functional Partners of MAMs Are Involved in AD
2.1. Mitofusins: MFNs
Mitofusins (MFN1 and MFN2) belongs to GTPases family and are located in the OMM. Both MFNs are involved in mitochondrial fusion process by forming homotypic and heterotypic interactions with the inner mitochondria membrane’s (IMM) OPA1 protein [31][32]. Besides, several studies proposed that MFN2 modulates mitochondrial membrane potential (ΔΨm) through the regulation of nuclear-encoded subunits of OXPHOS complexes I, II, III, and V [33]. MFN2 acts as a regulator of apoptosis in a cell type- and age-specific manner [34][35], and is linked to mitophagy [36]. The importance of MFN2 in organelles juxtaposition and the communication function of MAMs has been also proven through the regulation of mitochondria Ca2+ uptake [37]. MFN2 was also shown to be involved in dendritic outgrowth, spine formation in mice models, and to prevent neurodegeneration in the cerebellum through fusion mechanism [38].
Several studies reported that MFN2 is associated with AD [39] (Figure 2 and Table 1). MFN2 gene polymorphisms were reported in LOAD [40]. Moreover, the mitochondrial fusion and fission balance is altered in AD, correlating with enhanced or reduced MFNs expression [41]. A reduction of both MFN1 and MFN2 expression was reported in human AD brains [41][42]. Accordingly, a reduction of MFN2 expression was also reported in N2a cells expressing the APP familial Swedish mutation (APPswe) that overproduces Aβo [43]. On the contrary, an overexpression of MFN2 was observed in human cybrid cells in which mitochondria from mild cognitive impairment (MCI) subjects were incorporated into neuronal cells depleted of endogenous mitochondrial DNA [44]. These discrapencies may point out a complex regulation of MFN2 in AD pathogenesis. In fact, directly linked to APP processing in AD, MFN2 was shown to control γ-secretase activity and Aβ production by modulating ER and mitochondrial membrane contacts, without affecting BACE1 and neprilysin expression [45]. Moreover, PS2-linked FAD mutants promote ER-mitochondria coupling in a MFN2-dependent manner (Table 1) [46]. Altogether, these studies highlight a central role of MFN2 in AD pathogenesis through the regulation of MAMs tethering, mitochondrial structure and function, and cell death but also APP processing.
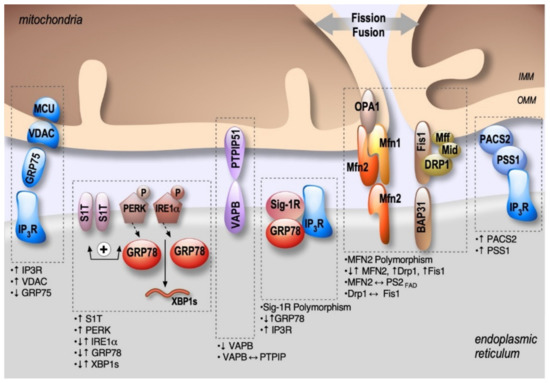
Figure 2. Schema showing the localization and alterations of MAMs molecular components in AD, including upregulation (↑) or down-regulation (↓) of MAMs proteins expression, MAMs proteins interactions (⟷), and genes polymorphism.
2.2. IP3R-Grp75-VDAC
The inositol 1,4,5-trisphosphate receptors (IP3Rs), releasing Ca2+ from the ER to the cytosol [47], are enriched in ER-mitochondria contact sites playing a key role in cellular differentiation, survival, and apoptosis [48][49]. Ca2+ uptake by mitochondria occurs through VDAC, an OMM protein which is a permeable Ca2+ channel, together with the mitochondrial Ca2+ uniporter (MCU) complex [50] located on the IMM (Figure 1). In MAMs, a functional complex between VDAC (isoform 1) and IP3R (isoform 3) is formed through a chaperone called glucose-regulated protein 75 (Grp75, a member of the heat shock protein 70 family) [51]. The IP3R-Grp75-VDAC complex regulates Ca²+ transfer from the ER to mitochondria, suggesting its involvement in Ca²+ homeostasis deregulation largely described in AD [51]. While there is only one study documenting a reduction of Grp75 expression in the temporal and parietal cortex of AD post-mortem brains [52], several studies demonstrated alterations of VDAC expression in AD (Figure 2 and Table 1). VDAC1 expression increases progressively according to AD stages in the cortex of post-mortem brains, in aged APP mice, and also in neuroblastoma cells treated with Aβo [53]. Accordingly, another study reported that Aβ treatment of primary hippocampal neurons increased VDAC1 and IP3R expression, resulting in enhanced contacts between ER and mitochondria [54]. VDAC1 also interacts with Aβ and pTau, likely contributing to mitochondrial dysfunction [55]. In turn, Aβ triggers VDAC dephosphorylation in lipid rafts in AD brains correlating with cell death [56]. Inversely, reduced expression of VDAC1 decreased the mRNA levels of APP, Tau, PS1 and PS2, and also BACE1 [57].
Altearations of IP3Rs expressions and activity have been largely explored in AD and were demonstrated to be linked to both Aβ and PS1 and PS2 FAD mutations. Fibroblasts from FAD patients exhibited an enhanced response to bombesin- and bradykinin-mediated IP3 generation in comparison to control cells [58]. These data were further confirmed in cortical and hippocampal neurons from 3xTgAD mice [59]. Aβo are also involved in IP3R overactivation by increasing IP3 production and triggering cytotoxicity [60]. Importantly, the reduction of IP3R1-mediated Ca2+ release in the cortical and hippocampal neurons of PS1M146V and 3xTgAD mice reduces ryanodine receptor (RyR)-mediated Ca2+ release, restores cAMP response element-binding (CREB)-dependent gene expression, rescues long-term potentiation (LTP) to normal levels, attenuates Aβ accumulation and pTau and reverses memory deficits [61]. Another study also showed a physical interaction of IP3R with PS1 (M146L) and PS2 (N141I) mutants stimulating IP3R gating activity [62] (Figure 2 and Table 1). Of note, our laboratory and others reported a major role of enhanced RyR expression and activity in AD [63][64][65][66][67][68]. Nevertheless, dedicated studies are still needed to demonstrate if the interaction between IP3R and FAD PSs mutants occurs preferentially in MAMs and to unravel the physiopathological contribution of RyRs in MAMs dysfunction related to AD.
2.3. PACS2-PSS1
Phosphofurin acidic cluster sorting protein 2 (PACS2) is a multifunctional sorting protein that interacts with several cargo proteins and regulates their location in MAMs [69]. PACS2 controls apoptosis induction, through the translocation of Bim (Bcl-2-like protein 11) to lysosomes [70], or Bid (BH3 interacting domain) to mitochondria [71]. The precise localization of PACS2 in MAMs is still uncertain. However, it has been demonstrated that PACS2 controls ER-mitochondrial apposition through its tethering action and is also involved in Ca2+ transfer from ER to mitochondria through IP3R localization [71]. Accordingly, PACS2 deletion leads to decreased ER-mitochondria contacts and triggers mitochondria fragmentation [71] and apoptosis [72].
PSS1 (phosphatidylserine synthase-1) is synthesized and specifically located in MAMs, supporting the direct transfer of lipids between ER and mitochondria [73]. PACS2 and PSS1 are functionally linked, since the overexpression of PACS2 raises the levels of PSS1 in MAMs [71].
PACS2 and PSS1 regulations in AD were reported in a study by Hedskog et al. showing enhanced PACS2 and PSS1 protein levels in AD transgenic mice expressing Swe/LDN FAD mutations [54]. More precisely, PACS2 protein expression was enhanced in the hippocampus of 2 month-old aged mice before Aβ plaque formation, and in the cortex and the cerebellum at 6 months. Meanwhile, PSS1 protein expression was only enhanced in the cerebellum starting at 6 months. Importantly, the authors also revealed enhanced PSS1 and PACS2 protein levels in human SAD brains (Figure 2 and Table 1). Enhanced PSS1 and PACS2 protein expressions were not corroborated by an increase in PSS1 and PACS2 mRNAs levels, suggesting that the regulation of PSS1 and PACS2 expressions in AD occurs at the protein level only. The authors also reported unchanged phospholipid metabolism in both mice and AD brains, thus questioning the functional consequences related to enhanced PSS1 and PACS-2 protein expression. Nevertheless, ablation of PSS2 triggers degeneration of astrocytes and neuronal hippocampal primary cultures. In conclusion, this unique study unravels the potential contribution of an additional MAMs molecular component in AD [54].
2.4. BAP31-FIS1-DRP1
Another physical tether of MAMs has been described as the interaction between the resident integral ER protein B cell receptor–associated protein 31 (BAP31) and the OMM mitochondrial FIS1 protein involved in the initiation of mitochondrial fission process [74][75]. FIS1 interacts with dynamin-related protein1 (DRP1) after its recruitment at mitochondrial fission sites by the OMM receptors (Mff, Mid49 and Mid51) in the MAMs complex, including BAP31 [76]. Intriguingly, a recent study reported that the human FIS1 protein can cause mitochondrial fragmentation in the absence of DRP1 and Dyn2 GTPases through its ability to bind to MFN1, MFN2, and OPA1, also inhibiting the mitochondrial fusion [77]. Shen et al. also demonstrated that mutations in the FIS1 gene do not cause defects in mitochondrial fission in response to fission-inductor chemical treatments but trigger persisting large aggregates of autophagosomes [76]. Accordingly, FIS1 KO MEFs cells display a strongly impaired mitophagy [78]. Iwasawa et al. showed that FIS1 conveys an apoptosis signal from the mitochondria to the ER through its interaction with BAP31 at the ER and its cleavage into the pro-apoptotic p20 [74]. The latter triggers a Ca2+ transfer from the ER to the mitochondria leading to ΔΨm depolarization, hence priming cell death induction [74][75]. BAP31 is also essential for mitochondrial homeostasis as it stimulates the formation of the mitochondrial complex I [79].
Several studies reported that FIS1, BAP31, and DRP1 may contribute individually to AD physiopathology (Figure 2 and Table 1). It was first reported that the mRNA and protein levels of FIS1 and DRP1 are increased in the frontal cortex of patients with early, definite, and severe AD [42]. Joshi et al., reported later that Aβ42-treated neurons and fibroblasts from SAD or FAD patients feature an increased interaction between DRP1 and FIS1, which causes mitochondrial dysfunctions attested by increased mitochondrial fragmentation, oxidative stress, decreased ΔΨm and ATP levels, and increases in the activity of pro-apoptotic enzymes and Cyt c release [80]. Most importantly, inhibiting the DRP1-FIS1 interaction corrects these mitochondrial impairments, reduces in the 5xFAD mice AD model (that overexpress human APP and PS1 transgenes with a total of five AD-linked mutations) cognitive defects, Aβ40 and Aβ42 levels, and oxidative stress, and increases ATP levels [80]. In parallel, BAP31 knockdown in the hippocampus and the cerebral cortex of PS1 M146V mice results in an increased BACE1 protein level, exacerbating C99 accumulation and Aβ plaque formation [81]. Other studies showed that pTau and Aβ monomers and oligomers can also directly interact with Drp1 in AD late stages, likely contributing to abnormal mitochondrial dynamics [42][82]. Moreover, Aβ also triggers nitric oxide production, likely contributing to Drp1 S-nitrosylation-mediated mitochondrial fragmentation and neuronal damage [83] (Figure 2 and Table 1). Together, these studies support the involvement of the BAP31-FIS1-DRP1 complex in AD pathology.
2.5. VAPB-PTPIP51
The OMM protein, tyrosine phosphatase-interacting protein-51 (PTPIP51), was identified as a binding partner for the resident ER protein vesicle-associated membrane protein-associated protein-B (VAPB) [84]. As a physical tether of MAMs, VAPB has several functions, including vesicle trafficking and UPR [84]. It was then demonstrated that the loss of either VAPB or PTPIP51 perturbs the uptake of Ca2+ by mitochondria, decreasing ATP production [85] and stimulating the formation of autophagosomes [86]. Additionally, PTPIP51 can interact with the oxysterol-binding protein (OSBP)-related proteins ORP5 and ORP8 in MAMs [87], likely contributing to phosphatidylserine (PS) transport between ER and mitochondria.
VAPB P56S mutation is associated with dominantly inherited familial forms of type-8 amyotrophic lateral sclerosis (ALS) [88]. Moreover, TAR DNA binding protein 43 (TDP-43) and fused in sarcoma (FUS), two ALS/frontotemporal dementia (FTD)-associated proteins, were reported to activate the glycogen synthase kinase-3β (GSK-3β), which inhibits VAPB-PTPIP51 binding, ultimately resulting in disrupted Ca2+ homeostasis and reduced ATP production [85][89]. Strikingly, it has recently been observed that AD patients with granulovacuolar degeneration bodies (GVB) feature lower VAPB level [90] (Figure 2 and Table 1). Interestingly, AD is a pathology where TDP-43 and FUS accumulate in stress granules and GVB. Furthermore, GSK-3β is also overactivated in AD [91]. A recent study reports a reduction of VAPB and PTPIP51 interaction in the pyramidal cortex of patients with early/mid dementia, and a reduction of the expression of both VAPB and PTPIP51 in the cortex of AD patients at late-stages [92]. Together, these studies also suggest the involvement of VAPB-PTPIP51 physical/or functional alterations in AD pathophysiology.
2.6. Sig-1R-Grp78
Sigma non-opioid intracellular 1-receptor 1 (Sig-1R) is a chaperone protein present in ER lipid rafts that interacts and modulates different proteins in ER and plasma membrane [93]. Sig-1R is essential for MAMs stability [94], and is implicated in lipid synthesis and trafficking [95].
Grp78 (or BiP), a member of the heat shock protein 70 family, is a chaperone and an ER stress sensor [96]. Importantly, Grp78 is present in MAMs, where it folds the steroidogenic acute regulatory protein (StAR) transporting cholesterol for delivery to the OMM [97]. Under ER stress conditions, Grp78 dissociates from and activates three branches of the UPR: activating transcription factor 6 (ATF6), IRE1α, and PERK. The UPR generates an adaptive or a pro-apoptotic response according to the stress duration and/or intensity [98]. In aging, the pro-apoptotic response becomes more common than the adaptive pathway, probably due to a failure in chaperone systems [98]. Interestingly, the impairment in UPR, apoptosis, and accumulation of misfolded proteins are common features of AD and other neurodegenerative diseases [99]. Desipte this, there is still no consensus on the role of Grp78 in AD. While APP/PS1 mice and Aβ25-35 treated neurons presented increased Grp78 expression (reviewed in Reference [100]), another study in AD post-mortem brains with PS1 mutations showed a reduction in Grp78 mRNA levels [101]. Recently, a study demonstrated that the 5xFAD mice had no changes in Grp78 expression and other ER stress-related proteins [102]. These discrepancies could be linked to the pleiotropic functions regulated by and regulating Grp78 expression (Figure 2 and Table 1).
Sig-1R forms a complex with Grp78 in MAMs under resting conditions [103]. During ER stress, Sig-1R also dissociates from Grp78 and forms a complex with IP3R through ankyrin B to increase Ca2+ signaling from ER to the mitochondria, thereby enhancing ATP synthesis [104]. Besides, Sig-1R stimulates phospholipase C (PLC) that increases IP3 levels, resulting also in Ca2+ release from the ER [105]. The activation of Sig-1R is considered neuroprotective since Sig-1R silencing in hippocampal neurons results in Cyt c release, caspase-3 activation, and a deficit in spine formation. Remarkably, this mechanism is reversed by superoxide dismutase activation, showing that the reduction of Sig-1R increases the levels of ROS [106].
Interestingly, Sig-1R has been associated with a variety of neurodegenerative diseases, including AD [107][108]. Reduced Sig-1R binding sites were reported in AD post-mortem brains [109]. It has also been proposed that a polymorphism in Sig-1R (TT-P) correlates with the risk of developing AD [110]. This finding has been challenged in another study demonstrating that another Sig-1R polymorphism (Q2P) may worsen clinical outcomes only in association with the gentic risk factor APOEε(epsilon)4 [111]. The protective effect of Sig-1R has also been observed in two AD mice models (3xTg-AD and MacGill-R-Thy1-APP), where the treatment with a Sig-1R agonist, named AF710B (a concomitant agonist of the muscarinic 1 receptor), have been shown to restore cognitive impairment, and to reduce amyloid pathology and neuroinflammation [112][113]. Accordingly, the silencing of Sig-1R or application of the Sig-1R antagonist (NE-100) aggravated the pathological status in Aβ25-35 treated mice (i.e., learning deficits, reduced BDNF levels, and enhanced expression of Bax) [114]. The treatment of Aβ25-35 injected mice with ANAVEX2-73, another agonist of Sig-1R, decreases pTau, Bax/Bcl-2 ratio, lipid peroxidation, C99 accumulation, and Cyt c release [115][116], while decreasing Sig-1R expression enhances Tau hyperphosphorylation and reduces dendritic spines formation [117].
Overall, these studies converge to demonstrate the neuroprotective effect of Sig-1R agonists, acting through a diversity of Sig-1R-mediated signaling functions that are generally pro-survival and anti-apoptotic.
2.7. Protein Kinase-Like Endoplasmic Reticulum Kinase: PERK
Several studies reported that the activation of the PERK pathway attenuates protein synthesis until the unfolded protein accumulation is removed [98]. Interestingly, PERK is enriched at MAMs [118], where it facilitates the tethering of the ER to mitochondria and sensitizes cells to apoptosis [118][119]. PERK KO cells show reduced MAMs and disturbed cytosolic Ca2+ signaling and also protect cells from ROS-induced mitochondrial dysfunction [118]. A recent study demonstrates that PERK localized in MAMs phosphorylates mitochondrial E3 ubiquitin ligases and decreases the formation of mitochondria-associated ER membranes under ER stress condition [120]. UPR occurs relatively early in AD, and PERK was shown to be activated in human AD-derived brains [121][122]. The dysregulated PERK pathway has been recapitulated in AD mice models that develop β-amyloidosis [123][124][125], Tau-mediated neurodegeneration [126][127], and in SAD genetic risk factor APOEε4 mice models [128] (Figure 2 and Table 1). Several studies have provided mechanistic insights into the direct relationship between PERK overactivation and several paradigms occurring in AD. The phosphorylation of eIF2α, occurring down-stream of PERK activation, has been shown to increase BACE1 expression, thereby enhancing the production of Aβ in neurons [129], and genetic reduction of PERK reduces BACE1 protein level and Aβ production in the 5xFAD mouse model [123]. Accordingly, the selective ablation of PERK improved the synaptic plasticity and spatial memory in mice harboring APP and PS1 mutations [124], consistent with the requirement for active protein translation in memory consolidation [130]. Another study demonstrated that a reduction in eIF2α phosphorylation enhances the late phase of LTP and memory in mice [131]. Furthermore, the local expression of ATF4 (a transcription factor activated downstream of PERK and eIF2α ER stress responsive pathway) in axons induces axonal damage through a cell-non-autonomous mechanism that propagates between neurons [132]. These data suggest that sustained PERK-eIF2α-ATF4 activation worsens AD pathogenesis, and that PERK localization in MAMs may act as a new pathogenic route contributing to AD development.
Table 1. MAMs molecular component alterations in AD. These alterations include reduced (↓), increased (↑), unchanged expressions (=), or interaction (⟷) between proteins observed in AD study models and human-derived samples (SAD brains and SAD/ FAD fibroblasts); KO: knock-out; KI: knock-in; “OE”: overexpressing; MCI: mild cognitive impairment; GVB: granulovacuolar degeneration bodies; Aβo: Aβ oligomers.
|
Proteins |
Alterations |
Study Models |
References |
|
MFNs |
Polymorphism |
AD patients |
[40] |
|
|
↓MFN2 & MFN1 (proteins and mRNA) |
AD brains (hippocampus & frontal cortex) & N2a “OE” APPswe |
|
|
|
↑MFN2 |
MCI cytoplasmic hybrid (cybrid) cells |
[44] |
|
|
↑MFN2⟷PS2 (FAD), ↑MAMs |
MFN2 KO MEFs FAD PS2 (N141I) Mice |
[46] |
|
IP3R-Grp75-VDAC |
↑VDAC Aβ⟷VDAC pTau⟷VDAC |
Tg2576 and J20 mice APP, APP/PS1, 3xTgAD mice & AD brains (cortical tissues) |
[53] [55] |
|
|
↑VDAC, ↑IP3R activity, ↑MAMs |
Ab-treated hippocampal neurons |
[54] |
|
|
VDAC dephosphorylation |
AD brains |
[56] |
|
|
↓Grp75 |
AD brains (temporal and parietal cortex) |
[52] |
|
|
FAD PS1/2⟷IP3R |
Sf9 cells |
[62] |
|
PACS2-PSS1 |
↑PACS2, ↑PSS1 |
APPswe/LDN mice AD brains cortex |
[54] |
|
BAP31-FIS1-DRP1 |
↑Fis1, ↑Drp1 |
AD brains (frontal cortex) |
[42] |
|
|
↑Drp1⟷Fis |
Aβ42-treated neurons, SAD/FAD fibroblasts, N2a “OE” APPswe, & 5xTgAD mice |
[80] |
|
|
pTau⟷Drp1 ↓Drp1 mito localization |
AD brains, APP, APP/PS1 & 3xTgAD mice Drosophila “OE” Tau R406W |
[55] [133] |
|
|
Ab⟷Drp1 |
AD brains (frontal cortex) |
[42] |
|
VAPB-PTPIP51 |
↓VAPB, ↓PTPIP51 ↓VAPB⟷PTPIP51 |
AD brains (subicular with GVB neurons) AD brains (cortex) |
[90] [92] |
|
Sig-1R-Grp78 |
↓Sig-1R Sig-1R polymorphism |
AD brains AD patients |
[109] |
|
|
↑Grp78 |
Streptozotocin model, APP/PS1 mice & Aβ25-35-treated rats |
|
|
|
↓Grp78 mRNA |
AD brains (FAD PS1) |
[101] |
|
|
=Grp78 expression |
5xFAD mice |
[102] |
|
PERK |
↑PERK pathway |
AD brains 5xFAD mice APP/PS1, APP(SL)/PS1 KI & rTg4510 mice APOE4 mice |
[121][122][123][124][125][126] [123] |
|
IRE1a |
↑↓IRE1a pathway, ↑↓XBP1s |
AD brains & 5xFAD mice Aβ42o-treated SH-SY5Y & CHO “OE” APPLDN |
[137] [138] |
|
S1T |
↑S1T
|
AD brains, SHSY-5Y “OE”APPswe or treated with Aβo |
[139]
|
2.8. Active Inositol-Requiring Transmembrane Kinase and Endonuclease Alpha: IRE1α
The UPR IRE1α branch increases protein folding trough the induction of chaperones and enhances the removal of misfolded proteins [98]. Active IRE1α processes the mRNA encoding X-box-binding protein 1 spliced isoform (XBP1s), a transcription factor that upregulates genes encoding mediators of protein folding, ERAD (ER-associated degradation), organelle biogenesis, and protein quality control [98]. A fraction of IRE1α is located at MAMs through Sig-1R stabilization responding to mitochondrial ROS [103][140]. A recent study demonstrated that the mitochondrial ubiquitin ligase (MITOL/MARCH5) inhibits ER stress-induced apoptosis through ubiquitylation and degradation of IRE1α at MAMs [141]. IRE1α at MAMs can also induce cell death via mitochondrial Ca2+ overload [142]. Another study linked IRE1α to mitochondrial dysfunction and apoptosis through the phosphorylation of ASK1 and JNK [143][144].
The activation of the IRE1α branch is implicated in AD pathogenesis, mainly through the activation of XBP1s [121][122] (Figure 2 and Table 1). Enhanced or reduced XBP1 mRNA splicing was reported in AD human brains [145]. XBP1s activation occurs upon Aβo treatment lowering BACE1 protein level [138] and activating Kalirin-7 (Kal7), a protein that controls synaptic plasticity [146]. This suggest that XBP1s acts as a protective mechanism in AD. Accordingly, the overexpression of XBP1s protects against Aβ toxicity [147] and pathological Tau [148]. In addition, a genomic screen identified a cluster of AD genes as possible direct targets of XBP1s, including APP, and components of the α-secretase, and proteins involved in APP trafficking and maturation [149]. Finally, in both Drosophila and mammalian cell culture models of AD, XBP1 overexpression also down-regulates RyR3 expression, which in turn prevents cytosolic Ca2+ overload [147]. These last data corroborate studies demonstrating that pharmacological and genetic approaches reducing RyR-mediated exacerbated ER Ca2+ leak provide protective effects and rescue several AD pathogenic paradigms in cellular and mice AD models [64][65][66].
2.9. The Truncated Variant of the Sarco-Endoplasmic Reticulum Ca2+-ATPase 1: S1T
S1T is among molecular partners at the ER-mitochondria interface regulating inter-organelle Ca2+ signaling and cell death [150][151][152]. We reported the induction of S1T expression under pathological ER stress conditions occurring through the PERK-eIF2α-ATF4-CHOP (C/EBP homologous protein) pathway. On the one hand, S1T induction amplifies ER stress response, and on the other hand, it enhances ER Ca2+ leak, increases the number of ER-mitochondria contact sites, and inhibits mitochondrial movements. This leads to increased Ca2+ transfer to mitochondria, thus activating the mitochondrial apoptotic pathway [152]. Interestingly, we recently reported a molecular interplay between S1T-dependent ER Ca2+ leak, UPR, and APP processing, likely contributing to AD pathogenesis [139] (Figure 2 and Table 1). We demonstrated that S1T expression is increased in SAD brains and correlates with Aβ and ER stress chaperone protein levels (Grp78 and Calreticulin). Increased S1T expression is induced in an Aβ-dependent manner and enhances in return the production of APP-CTFs and Aβ through specific increases of BACE1 expression and activity. Interestingly, the induction of S1T expression is also linked to neuroinflammation [139]. Other studies are ongoing to unravel the contribution of S1T-mediated ER Ca2+ leak to MAMs dysfunctions, synaptic plasticity alterations, and cognitive deficits in AD.
References
- Checler, F. Processing of the beta-amyloid precursor protein and its regulation in Alzheimer’s disease. J. Neurochem. 1995, 65, 1431–1444.
- Nhan, H.S.; Chiang, K.; Koo, E.H. The multifaceted nature of amyloid precursor protein and its proteolytic fragments: Friends and foes. Acta Neuropathol. 2015, 129, 1–19, doi:10.1007/s00401-014-1347-2.
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923, doi:10.1126/science.8346443.
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-Analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458, doi:10.1038/ng.2802.
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430, doi:10.1038/s41588-019-0358-2.
- Bellenguez, C.; Grenier-Boley, B.; Lambert, J.C. Genetics of Alzheimer’s disease: Where we are, and where we are going. Curr. Opin. Neurobiol. 2020, 61, 40–48, doi:10.1016/j.conb.2019.11.024.
- Tanzi, R.E.; Bertram, L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell 2005, 120, 545–555, doi:10.1016/j.cell.2005.02.008.
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430, doi:10.1038/gim.2015.117.
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185, doi:10.1126/science.1566067.
- Panza, F.; Lozupone, M.; Logroscino, G.; Imbimbo, B.P. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2019, 15, 73–88, doi:10.1038/s41582-018-0116-6.
- Flammang, B.; Pardossi-Piquard, R.; Sevalle, J.; Debayle, D.; Dabert-Gay, A.S.; Thévenet, A.; Lauritzen, I.; Checler, F. Evidence that the amyloid-β protein precursor intracellular domain, AICD, derives from β-secretase-generated C-terminal fragment. J. Alzheimer’s Dis. 2012, 30, 145–153, doi:10.3233/JAD-2012-112186.
- Willem, M.; Tahirovic, S.; Busche, M.A.; Ovsepian, S.V.; Chafai, M.; Kootar, S.; Hornburg, D.; Evans, L.D.B.; Moore, S.; Daria, A.; et al. n-Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature 2015, 526, 443–447, doi:10.1038/nature14864.
- Zhang, Z.; Song, M.; Liu, X.; Su Kang, S.; Duong, D.M.; Seyfried, N.T.; Cao, X.; Cheng, L.; Sun, Y.E.; Ping Yu, S.; et al. Delta-secretase cleaves amyloid precursor protein and regulates the pathogenesis in Alzheimer’s disease. Nat. Commun. 2015, 6, 1–16, doi:10.1038/ncomms9762.
- Baranger, K.; Marchalant, Y.; Bonnet, A.E.; Crouzin, N.; Carrete, A.; Paumier, J.M.; Py, N.A.; Bernard, A.; Bauer, C.; Charrat, E.; et al. MT5-MMP is a new pro-amyloidogenic proteinase that promotes amyloid pathology and cognitive decline in a transgenic mouse model of Alzheimer’s disease. Cell. Mol. Life Sci. 2016, 73, 217–236, doi:10.1007/s00018-015-1992-1.
- Bourgeois, A.; Lauritzen, I.; Lorivel, T.; Bauer, C.; Checler, F.; Pardossi-Piquard, R. Intraneuronal accumulation of C99 contributes to synaptic alterations, apathy-like behavior, and spatial learning deficits in 3 × TgAD and 2 × TgAD mice. Neurobiol. Aging 2018, 71, 21–31, doi:10.1016/j.neurobiolaging.2018.06.038.
- Lauritzen, I.; Pardossi-Piquard, R.; Bauer, C.; Brigham, E.; Abraham, J.D.; Ranaldi, S.; Fraser, P.; St-George-Hyslop, P.; Le Thuc, O.; Espin, V.; et al. The β-secretase-derived C-terminal fragment of βAPP, C99, but not Aβ, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J. Neurosci. 2012, 32, 16243–16255, doi:10.1523/JNEUROSCI.2775-12.2012.
- Lauritzen, I.; Pardossi-Piquard, R.; Bourgeois, A.; Pagnotta, S.; Biferi, M.G.; Barkats, M.; Lacor, P.; Klein, W.; Bauer, C.; Checler, F. Intraneuronal aggregation of the β-CTF fragment of APP (C99) induces Aβ-independent lysosomal-autophagic pathology. Acta Neuropathol. 2016, 132, 257–276.
- Kwart, D.; Gregg, A.; Scheckel, C.; Murphy, E.A.; Paquet, D.; Duffield, M.; Fak, J.; Olsen, O.; Darnell, R.B.; Tessier-Lavigne, M. Erratum: A large panel of isogenic APP and PSEN1 mutant human iPSC neurons reveals shared endosomal abnormalities mediated by APP β-CTFs, not Aβ. Neuron 2019, 104, 256–270, doi:10.1016/j.neuron.2019.11.010.
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium 2002, 32, 235–249, doi:10.1016/S0143416002001823.
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723, doi:10.1016/j.beem.2012.05.003.
- Giacomello, M.; Pellegrini, L. The coming of age of the mitochondria—ER contact: A matter of thickness. 2016, 23, 1417–1427, doi:10.1038/cdd.2016.52.
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766, doi:10.1126/science.280.5370.1763.
- Csordás, G.; Renken, C.; Várnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnóczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921, doi:10.1083/jcb.200604016.
- Walter, L.; Hajnóczky, G. Mitochondria and endoplasmic reticulum: The lethal interorganelle cross-talk. J. Bioenerg. Biomembr. 2005, 37, 191–206.
- Veeresh, P.; Kaur, H.; Sarmah, D.; Mounica, L.; Verma, G.; Kotian, V.; Kesharwani, R.; Kalia, K.; Borah, A.; Wang, X.; et al. Endoplasmic reticulum–mitochondria crosstalk: From junction to function across neurological disorders. Ann. N. Y. Acad. Sci. 2019, 1457, 41–60, doi:10.1111/nyas.14212.
- van Vliet, A.R.; Agostinis, P. Mitochondria-Associated membranes and ER stress. In Current Topics in Microbiology and Immunology; Springer Nature Switzerland AG, 2018; Volume 414, pp. 73–102.
- Gerakis, Y.; Hetz, C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 2018, 285, 995–1011, doi:10.1111/febs.14332.
- Hashimoto, S.; Saido, T.C. Critical review: Involvement of endoplasmic reticulum stress in the aetiology of Alzheimer’s disease. Open Biol. 2018, 8, doi:10.1098/rsob.180024.
- Esteras, N.; Abramov, A.Y. Mitochondrial calcium deregulation in the mechanism of beta-amyloid and tau pathology. Cells 2020, 9, doi:10.3390/cells9092135.
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 1–22, doi:10.1186/s13024-020-00376-6.
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005, 280, 26185–26192, doi:10.1074/jbc.M503062200.
- Cipolat, S.; De Brito, O.M.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932, doi:10.1073/pnas.0407043101.
- Pich, S.; Bach, D.; Briones, P.; Liesa, M.; Camps, M.; Testar, X.; Palacín, M.; Zorzano, A. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum. Mol. Genet. 2005, 14, 1405–1415, doi:10.1093/hmg/ddi149.
- Youle, R.J.; Karbowski, M. Mitochondrial fission in apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 657–663, doi:10.1038/nrm1697.
- Martorell‐Riera, A.; Segarra‐Mondejar, M.; Muñoz, J.P.; Ginet, V.; Olloquequi, J.; Pérez‐Clausell, J.; Palacín, M.; Reina, M.; Puyal, J.; Zorzano, A.; et al. Mfn2 downregulation in excitotoxicity causes mitochondrial dysfunction and delayed neuronal death. EMBO J. 2014, 33, 2388–2407, doi:10.15252/embj.201488327.
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380, doi:10.1083/jcb.201007013.
- Naon, D.; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernández-Alvarez, M.I.; et al. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254, doi:10.1073/pnas.1606786113.
- Chen, H.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 2007, 130, 548–562, doi:10.1016/j.cell.2007.06.026.
- Zhang, L.; Trushin, S.; Christensen, T.A.; Bachmeier, B.V.; Gateno, B.; Schroeder, A.; Yao, J.; Itoh, K.; Sesaki, H.; Poon, W.W.; et al. Altered brain energetics induces mitochondrial fission arrest in Alzheimer’s Disease. Sci. Rep. 2016, 6, 1–12, doi:10.1038/srep18725.
- Kim, Y.J.; Park, J.K.; Kang, W.S.; Kim, S.K.; Han, C.; Na, H.R.; Park, H.J.; Kim, J.W.; Kim, Y.Y.; Park, M.H.; et al. Association between mitofusin 2 gene polymorphisms and late-onset Alzheimer’s disease in the Korean population. Psychiatry Investig. 2017, 14, 81–85, doi:10.4306/pi.2017.14.1.81.
- Wang, X.; Su, B.; Lee, H.G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 2009, 29, 9090–9103, doi:10.1523/JNEUROSCI.1357-09.2009.
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509, doi:10.1093/hmg/ddr139.
- Park, J.; Choi, H.; Min, J.S.; Kim, B.; Lee, S.R.; Yun, J.W.; Choi, M.S.; Chang, K.T.; Lee, D.S. Loss of mitofusin 2 links beta-amyloid-mediated mitochondrial fragmentation and Cdk5-induced oxidative stress in neuron cells. J. Neurochem. 2015, 132, 687–702, doi:10.1111/jnc.12984.
- Gan, X.; Wu, L.; Huang, S.; Zhong, C.; Li, G.; Yu, H.; Swerdlow, R.H.; Chen, J.X.; Yan, S.S. Oxidative stress-mediated activation of extracellualr signal-regulated kinase contributes to mild cognitive impairment-related mitochondrial dysfuntion. Free Radic. Biol. Med. 2014, 75, 230–240, doi:10.1016/j.physbeh.2017.03.040.
- Leal, N.S.; Schreiner, B.; Pinho, C.M.; Filadi, R.; Wiehager, B.; Karlström, H.; Pizzo, P.; Ankarcrona, M. Mitofusin-2 knockdown increases ER-mitochondria contact and decreases amyloid β-peptide production. J. Cell. Mol. Med. 2016, 20, 1686–1695, doi:10.1111/jcmm.12863.
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum-mitochondria coupling by tuning the antagonistic effect of mitofusin 2. Cell Rep. 2016, 15, 2226–2238, doi:10.1016/j.celrep.2016.05.013.
- Berridge, M.J. Inositol triphosphate and calcium signalling. Nature 1993, 361, 315–325.
- Joseph, S.K.; Hajnóczky, G. IP3 receptors in cell survival and apoptosis: Ca2+ release and beyond. Apoptosis 2007, 12, 951–968, doi:10.1007/s10495-007-0719-7.
- Mikoshiba, K. IP3 receptor/Ca2+ channel: From discovery to new signaling concepts. J. Neurochem. 2007, 102, 1426–1446, doi:10.1111/j.1471-4159.2007.04825.x.
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340, doi:10.1038/nature10230.
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-Mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911, doi:10.1083/jcb.200608073.
- Yoo, B.C.; Kim, S.H.; Cairns, N.; Fountoulakis, M.; Lubec, G. Deranged expression of molecular chaperones in brains of patients with Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2001, 280, 249–258, doi:10.1006/bbrc.2000.4109.
- Cuadrado-Tejedor, M.; Vilariño, M.; Cabodevilla, F.; Del Río, J.; Frechilla, D.; Pérez-Mediavilla, A. Enhanced expression of the voltage-dependent anion channel 1 (VDAC1) in Alzheimer’s disease transgenic mice: An insight into the pathogenic effects of amyloid-β. J. Alzheimers Dis. 2011, 23, 195–206, doi:10.3233/JAD-2010-100966.
- Hedskog, L.; Pinho, C.M.; Filadi, R.; Rönnbäck, A.; Hertwig, L.; Wiehager, B.; Larssen, P.; Gellhaar, S.; Sandebring, A.; Westerlund, M.; et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. USA 2013, 110, 7916–7921, doi:10.1073/pnas.1300677110.
- Manczak, M.; Reddy, P.H. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 5131–5146, doi:10.1093/hmg/dds360.
- Fernandez-Echevarria, C.; Díaz, M.; Ferrer, I.; Canerina-Amaro, A.; Marin, R. Aβ promotes VDAC1 channel dephosphorylation in neuronal lipid rafts. Relevance to the mechanisms of neurotoxicity in Alzheimer’s disease. Neuroscience 2014, 278, 354–366, doi:10.1016/j.neuroscience.2014.07.079.
- Manczak, M.; Sheiko, T.; Craigen, W.J.; Reddy, P.H. Reduced VDAC1 protects against Alzheimer’s disease, mitochondria, and synaptic deficiencies. J. Alzheimer’s Dis. 2013, 37, 679–690, doi:10.3233/JAD-130761.
- Ito, E.; Oka, K.; Etcheberrigaray, R.; Nelson, T.J.; McPhie, D.L.; Tofel-Grehl, B.; Gibson, G.E.; Alkon, D.L. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 534–538, doi:10.1073/pnas.91.2.534.
- Cheung, K.; Mei, L.; Mak, D.D.; Hayashi, I.; Iwatsubo, T.; Kang, D.E.; Foskett, J.K. Gain-of-function enhancement of InsP3 receptor modal gating by familial Alzheimer’s disease-linked presenilin mutants in human cells and mouse neurons. Sci. Signal. 2010, 3, doi:10.1126/scisignal.2000818.Gain-of-function.
- Demuro, A.; Parker, I. Cytotoxicity of intracellular Aβ42 amyloid oligomers involves Ca2+ release from the endoplasmic reticulum by stimulated production of inositol trisphosphate. J. Neurosci. 2013, 33, 3824–3833.
- Shilling, D.; Müller, M.; Takano, H.; Mak, D.O.D.; Abel, T.; Coulter, D.A.; Foskett, J.K. Suppression of InsP3 receptor-mediated Ca2+ signaling alleviates mutant presenilin-linked familial Alzheimer’s disease pathogenesis. J. Neurosci. 2014, 34, 6910–6923, doi:10.1523/JNEUROSCI.5441-13.2014.
- Cheung, K.H.; Shineman, D.; Müller, M.; Cárdenas, C.; Mei, L.; Yang, J.; Tomita, T.; Iwatsubo, T.; Lee, V.M.Y.; Foskett, J.K. Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron 2008, 58, 871–883, doi:10.1016/j.neuron.2008.04.015.
- Del Prete, D.; Checler, F.; Chami, M. Ryanodine receptors: physiological function and deregulation in Alzheimer disease. Mol. Neurodegener. 2014, 9, 21, doi:10.1186/1750-1326-9-21.
- Lacampagne, A.; Liu, X.; Reiken, S.; Bussiere, R.; Meli, A.C.; Lauritzen, I.; Teich, A.F.; Zalk, R.; Saint, N.; Arancio, O.; et al. Post-Translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer’s disease-like pathologies and cognitive deficits. Acta Neuropathol. 2017, 134, 749–767, doi:10.1007/s00401-017-1733-7.
- Bussiere, R.; Lacampagne, A.; Reiken, S.; Liu, X.; Scheuerman, V.; Zalk, R.; Martin, C.; Checler, F.; Marks, A.R.; Chami, M. Amyloid beta production is regulated by beta2-adrenergic signaling-mediated post-translational modifications of the ryanodine receptor. J. Biol. Chem. 2017, 292, 10153–10168, doi:10.1074/jbc.M116.743070.
- Oules, B.; Del Prete, D.; Greco, B.; Zhang, X.; Lauritzen, I.; Sevalle, J.; Moreno, S.; Paterlini-Brechot, P.; Trebak, M.; Checler, F.; et al. Ryanodine receptor blockade reduces amyloid-beta load and memory impairments in Tg2576 mouse model of Alzheimer disease. J. Neurosci. 2012, 32, 11820–11834, doi:10.1523/JNEUROSCI.0875-12.2012.
- Stutzmann, G.E.; Smith, I.; Caccamo, A.; Oddo, S.; LaFerla, F.M.; Parker, I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J. Neurosci. 2006, 26, 5180–5189, doi:10.1523/JNEUROSCI.0739-06.2006.
- Stutzmann, G.E.; Smith, I.; Caccamo, A.; Oddo, S.; Parker, I.; Laferla, F. Enhanced ryanodine-mediated calcium release in mutant PS1-expressing Alzheimer’s mouse models. Ann. N. Y. Acad. Sci. 2007, 1097, 265–277, doi:10.1196/annals.1379.025.
- Li, C.; Li, L.; Yang, M.; Zeng, L.; Sun, L. PACS-2: A key regulator of mitochondria-associated membranes (MAMs). Pharmacol. Res. 2020, 160, 105080, doi:10.1016/j.phrs.2020.105080.
- Werneburg, N.W.; Bronk, S.F.; Guicciardi, M.E.; Thomas, L.; Dikeakos, J.D.; Thomas, G.; Gores, G.J. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) protein-induced lysosomal translocation of proapoptotic effectors is mediated by phosphofurin acidic cluster sorting protein-2 (PACS-2). J. Biol. Chem. 2012, 287, 24427–24437, doi:10.1074/jbc.M112.342238.
- Simmen, T.; Aslan, J.E.; Blagoveshchenskaya, A.D.; Thomas, L.; Wan, L.; Xiang, Y.; Feliciangeli, S.F.; Hung, C.-H.; Crump, C.M.; Thomas, G. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 2005, 24, 717–729, doi:10.1038/sj.emboj.7600559.
- Youker, R.T.; Shinde, U.; Day, R.; Thomas, G. At the crossroads of homoeostasis and disease: Roles of the PACS proteins in membrane traffic and apoptosis. Biochem. J. 2009, 421, 1–15, doi:10.1042/BJ20081016.
- Stone, S.J.; Vance, J.E. Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J. Biol. Chem. 2000, 275, 34534–34540, doi:10.1074/jbc.M002865200.
- Iwasawa, R.; Mahul-Mellier, A.-L.; Datler, C.; Pazarentzos, E.; Grimm, S. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 2011, 30, 556–568, doi:10.1038/emboj.2010.346.
- Pagliuso, A.; Cossart, P.; Stavru, F. The ever-growing complexity of the mitochondrial fission machinery. Cell. Mol. Life Sci. 2018, 75, 355–374, doi:10.1007/s00018-017-2603-0.
- Shen, Q.; Yamano, K.; Head, B.P.; Kawajiri, S.; Cheung, J.T.M.; Wang, C.; Cho, J.-H.; Hattori, N.; Youle, R.J.; van der Bliek, A.M. Mutations in Fis1 disrupt orderly disposal of defective mitochondria. Mol. Biol. Cell 2014, 25, 145–159, doi:10.1091/mbc.E13-09-0525.
- Yu, R.; Jin, S.; Lendahl, U.; Nistér, M.; Zhao, J. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. 2019, 38, doi:10.15252/embj.201899748.
- Rojansky, R.; Cha, M.-Y.; Chan, D.C. Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on PARKIN and MUL1. Elife 2016, 5, doi:10.7554/eLife.17896.
- Namba, T. BAP31 regulates mitochondrial function via interaction with Tom40 within ER-mitochondria contact sites. Sci. Adv. 2019, 5, doi:10.1126/sciadv.aaw1386.
- Joshi, A.U.; Saw, N.L.; Shamloo, M.; Mochly-Rosen, D. Drp1/Fis1 interaction mediates mitochondrial dysfunction, bioenergetic failure and cognitive decline in Alzheimer’s disease. Oncotarget 2017, 9, 6128–6143, doi:10.18632/oncotarget.23640.
- Wang, T.; Chen, J.; Hou, Y.; Yu, Y.; Wang, B. BAP31 deficiency contributes to the formation of amyloid‐β plaques in Alzheimer’s disease by reducing the stability of RTN3. FASEB J. 2019, 33, 4936–4946, doi:10.1096/fj.201801702R.
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547, doi:10.1093/hmg/dds072.
- Cho, D.-H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-Nitrosylation of Drp1 Mediates β-Amyloid-Related Mitochondrial Fission and Neuronal Injury. Science 2009, 324, 102–105.
- De Vos, K.J.; Mórotz, G.M.; Stoica, R.; Tudor, E.L.; Lau, K.-F.; Ackerley, S.; Warley, A.; Shaw, C.E.; Miller, C.C.J. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 2012, 21, 1299–1311, doi:10.1093/hmg/ddr559.
- Stoica, R.; Paillusson, S.; Gomez‐Suaga, P.; Mitchell, J.C.; Lau, D.H.W.; Gray, E.H.; Sancho, R.M.; Vizcay‐Barrena, G.; De Vos, K.J.; Shaw, C.E.; et al. ALS/FTD‐associated FUS activates GSK‐3β to disrupt the VAPB–PTPIP51 interaction and ER–mitochondria associations. EMBO Rep. 2016, 17, 1326–1342, doi:10.15252/embr.201541726.
- Gomez-Suaga, P.; Paillusson, S.; Stoica, R.; Noble, W.; Hanger, D.P.; Miller, C.C.J. The ER-Mitochondria tethering complex VAPB-PTPIP51 regulates autophagy. Curr. Biol. 2017, 27, 371–385, doi:10.1016/j.cub.2016.12.038.
- Galmes, R.; Houcine, A.; Vliet, A.R.; Agostinis, P.; Jackson, C.L.; Giordano, F. ORP5/ORP8 localize to endoplasmic reticulum–mitochondria contacts and are involved in mitochondrial function. EMBO Rep. 2016, 17, 800–810, doi:10.15252/embr.201541108.
- Tokutake, Y.; Yamada, K.; Ohata, M.; Obayashi, Y.; Tsuchiya, M.; Yonekura, S. ALS-Linked P56S-VAPB mutation impairs the formation of multinuclear myotube in C2C12 Cells. Int. J. Mol. Sci. 2015, 16, 18628–18641, doi:10.3390/ijms160818628.
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.-F.; Vizcay-Barrena, G.; Lin, W.-L.; Xu, Y.-F.; Lewis, J.; et al. ER-Mitochondria associations are regulated by the VAPB–PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, doi:10.1038/ncomms4996.
- Yamoah, A.; Tripathi, P.; Sechi, A.; Köhler, C.; Guo, H.; Chandrasekar, A.; Nolte, K.W.; Wruck, C.J.; Katona, I.; Anink, J.; et al. Aggregates of RNA binding proteins and ER chaperones linked to exosomes in granulovacuolar degeneration of the Alzheimer’s disease brain. J. Alzheimer’s Dis. JAD 2020, 139–156, doi:10.3233/JAD-190722.
- Llorens-Marítin, M.; Jurado, J.; Hernández, F.; Ávila, J. GSK-3β, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7, doi:10.3389/fnmol.2014.00046.
- Lau, D.H.W.; Paillusson, S.; Hartopp, N.; Rupawala, H.; Mórotz, G.M.; Gomez-Suaga, P.; Greig, J.; Troakes, C.; Noble, W.; Miller, C.C.J. Disruption of endoplasmic reticulum-mitochondria tethering proteins in post-mortem Alzheimer’s disease brain. Neurobiol. Dis. 2020, 143, 105020, doi:10.1016/j.nbd.2020.105020.
- Hayashi, T.; Su, T.P. σ-1 receptors (σ1 binding sites) form raft-like microdomains and target lipid droplets on the endoplasmic reticulum: Roles in endoplasmic reticulum lipid compartmentalization and export. J. Pharmacol. Exp. Ther. 2003, 306, 718–725, doi:10.1124/jpet.103.051284.
- Watanabe, S.; Ilieva, H.; Tamada, H.; Nomura, H.; Komine, O.; Endo, F.; Jin, S.; Mancias, P.; Kiyama, H.; Yamanaka, K. Mitochondria‐associated membrane collapse is a common pathomechanism in SIGMAR 1 ‐ and SOD 1 ‐linked ALS . EMBO Mol. Med. 2016, 8, 1421–1437, doi:10.15252/emmm.201606403.
- Hayashi, T.; Su, T.P. The potential role of sigma-1 receptors in lipid transport and lipid raft reconstitution in the brain: Implication for drug abuse. Life Sci. 2005, 77, 1612–1624, doi:10.1016/j.lfs.2005.05.009.
- Gorbatyuk, M.; Gorbatyuk, O. The Molecular Chaperone GRP78/BiP as a Therapeutic Target for Neurodegenerative Disorders: A Mini Review. J. Genet. Syndr. Gene Ther. 2013, 4, doi:10.4172/2157-7412.1000128.
- Prasad, M.; Pawlak, K.J.; Burak, W.E.; Perry, E.E.; Marshall, B.; Whittal, R.M.; Bose, H.S. Mitochondrial metabolic regulation by GRP78. Sci. Adv. 2017, 3, doi:10.1126/sciadv.1602038.
- Hetz, C.; Papa, F.R. The unfolded protein response and cell fate control. Mol. Cell 2018, 69, 169–181, doi:10.1016/j.molcel.2017.06.017.
- Hoozemans, J.J.M.; van Haastert, E.S.; Nijholt, D.A.T.; Rozemuller, A.J.M.; Scheper, W. Activation of the unfolded protein response is an early event in Alzheimer’s and Parkinson’s disease. Neurodegener. Dis. 2012, 10, 212–215, doi:10.1159/000334536.
- Uddin, M.S.; Tewari, D.; Sharma, G.; Kabir, M.T.; Barreto, G.E.; Bin-Jumah, M.N.; Perveen, A.; Abdel-Daim, M.M.; Ashraf, G.M. Molecular mechanisms of ER stress and UPR in the pathogenesis of Alzheimer’s disease. Mol. Neurobiol. 2020, 57, 2902–2919, doi:10.1007/s12035-020-01929-y.
- Katayama, T.; Imaizumi, K.; Sato, N.; Miyoshi, K.; Kudo, T.; Hitomi, J.; Morihara, T.; Yoneda, T.; Gomi, F.; Mori, Y.; et al. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat. Cell Biol. 1999, 1, 479–485, doi:10.1038/70265.
- Sadleir, K.R.; Popovic, J.; Vassar, R. ER stress is not elevated in the 5XFAD mouse model of Alzheimer’s disease. J. Biol. Chem. 2018, 293, 18434–18443, doi:10.1074/jbc.RA118.005769.
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 2007, 131, 596–610, doi:10.1016/j.cell.2007.08.036.
- Hayashi, T.; Su, T.P. Regulating ankyrin dynamics: Roles of sigma-1 receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 491–496, doi:10.1073/pnas.98.2.491.
- Morin-Surun, M.P.; Collin, T.; Denavit-Saubie, M.; Baulieu, E.E.; Monnet, F.P. Intracellular σ1 receptor modulates phospholipase C and protein kinase C activities in the brainstem. Proc. Natl. Acad. Sci. USA 1999, 96, 8196–8199, doi:10.1073/pnas.96.14.8196.
- Tsai, S.Y.; Hayashi, T.; Harvey, B.K.; Wang, Y.; Wu, W.W.; Shen, R.F.; Zhang, Y.; Becker, K.G.; Hoffer, B.J.; Su, T.P. Sigma-1 receptors regulate hippocampal dendritic spine formation via a free radical-sensitive mechanism involving Rac1•GTP pathway. Proc. Natl. Acad. Sci. USA 2009, 106, 22468–22473, doi:10.1073/pnas.0909089106.
- Ryskamp, D.A.; Korban, S.; Zhemkov, V.; Kraskovskaya, N.; Bezprozvanny, I. Neuronal sigma-1 receptors: Signaling functions and protective roles in neurodegenerative diseases. Front. Neurosci. 2019, 13, 1–20, doi:10.3389/fnins.2019.00862.
- Delprat, B.; Crouzier, L.; Su, T.-P.; Maurice, T. At the crossing of ER stress and MAMs: A key role of Sigma-1 receptor? Adv. Exp. Med. Biol. 2020, 1131, 699–718, doi:10.1007/978-3-030-12457-1_28.
- Jansen, K.L.R.; Faull, R.L.M.; Storey, P.; Leslie, R.A. Loss of sigma binding sites in the CA1 area of the anterior hippocampus in Alzheimer’s disease correlates with CA1 pyramidal cell loss. Brain Res. 1993, 623, 299–302, doi:10.1016/0006-8993(93)91441-T.
- Fehér, Á.; Juhász, A.; László, A.; Kálmán, J.; Pákáski, M.; Kálmán, J.; Janka, Z. Association between a variant of the sigma-1 receptor gene and Alzheimer’s disease. Neurosci. Lett. 2012, 517, 136–139, doi:10.1016/j.neulet.2012.04.046.
- Huang, Y.; Zheng, L.; Halliday, G.; Dobson-Stone, C.; Wang, Y.; Tang, H.-D.; Cao, L.; Deng, Y.-L.; Wang, G.; Zhang, Y.-M.; et al. Genetic polymorphisms in Sigma-1 receptor and apolipoprotein E interact to influence the severity of Alzheimers disease. Curr. Alzheimer Res. 2011, 8, 765–770, doi:10.2174/156720511797633232.
- Fisher, A.; Bezprozvanny, I.; Wu, L.; Ryskamp, D.A.; Bar-Ner, N.; Natan, N.; Brandeis, R.; Elkon, H.; Nahum, V.; Gershonov, E.; et al. AF710B, a Novel M1/sigma1 agonist with therapeutic efficacy in animal models of Alzheimer’s disease. Neurodegener. Dis. 2016, 16, 95–110, doi:10.1159/000440864.
- Hall, H.; Iulita, M.F.; Gubert, P.; Flores Aguilar, L.; Ducatenzeiler, A.; Fisher, A.; Cuello, A.C. AF710B, an M1/sigma-1 receptor agonist with long-lasting disease-modifying properties in a transgenic rat model of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 811–823, doi:10.1016/j.jalz.2017.11.009.
- Maurice, T.; Strehaiano, M.; Duhr, F.; Chevallier, N. Amyloid toxicity is enhanced after pharmacological or genetic invalidation of the σ1 receptor. Behav. Brain Res. 2018, 339, 1–10, doi:10.1016/j.bbr.2017.11.010.
- Lahmy, V.; Meunier, J.; Malmström, S.; Naert, G.; Givalois, L.; Kim, S.H.; Villard, V.; Vamvakides, A.; Maurice, T. Blockade of tau hyperphosphorylation and aβ 1-42 generation by the aminotetrahydrofuran derivative ANAVEX2-73, a mixed muscarinic and σ 1 receptor agonist, in a nontransgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 2013, 38, 1706–1723, doi:10.1038/npp.2013.70.
- Lahmy, V.; Long, R.; Morin, D.; Villard, V.; Maurice, T. Mitochondrial protection by the mixed muscarinic/σ1 ligand ANAVEX2-73, a tetrahydrofuran derivative, in Aβ25–35 peptide-injected mice, a nontransgenic Alzheimer’s disease model. Front. Cell. Neurosci. 2015, 8, 1–11, doi:10.3389/fncel.2014.00463.
- Tsai, S.Y.A.; Pokrass, M.J.; Klauer, N.R.; Nohara, H.; Su, T.P. Sigma-1 receptor regulates Tau phosphorylation and axon extension by shaping p35 turnover via myristic acid. Proc. Natl. Acad. Sci. USA 2015, 112, 6742–6747, doi:10.1073/pnas.1422001112.
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891, doi:cdd201274 [pii]10.1038/cdd.2012.74.
- Munoz, J.P.; Ivanova, S.; Sanchez-Wandelmer, J.; Martinez-Cristobal, P.; Noguera, E.; Sancho, A.; Diaz-Ramos, A.; Hernandez-Alvarez, M.I.; Sebastian, D.; Mauvezin, C.; et al. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 2013, 32, 2348–2361, doi:10.1038/emboj.2013.168.
- Toyofuku, T.; Okamoto, Y.; Ishikawa, T.; Sasawatari, S.; Kumanogoh, A. LRRK2 regulates endoplasmic reticulum-mitochondrial tethering through the PERK-mediated ubiquitination pathway. EMBO J. 2020, 39, e100875, doi:10.15252/embj.2018100875.
- Hoozemans, J.J.; Veerhuis, R.; Van Haastert, E.S.; Rozemuller, J.M.; Baas, F.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 2005, 110, 165–172.
- Hoozemans, J.J.; van Haastert, E.S.; Nijholt, D.A.; Rozemuller, A.J.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am. J. Pathol. 2009, 174, 1241–1251, doi:10.2353/ajpath.2009.080814.
- Devi, L.; Ohno, M. PERK mediates eIF2alpha phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2272–2281, doi:10.1016/j.neurobiolaging.2014.04.031.
- Ma, T.; Trinh, M.A.; Wexler, A.J.; Bourbon, C.; Gatti, E.; Pierre, P.; Cavener, D.R.; Klann, E. Suppression of eIF2alpha kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat. Neurosci. 2013, 16, 1299–1305, doi:10.1038/nn.3486.
- Page, G.; Rioux Bilan, A.; Ingrand, S.; Lafay-Chebassier, C.; Pain, S.; Perault Pochat, M.C.; Bouras, C.; Bayer, T.; Hugon, J. Activated double-stranded RNA-dependent protein kinase and neuronal death in models of Alzheimer’s disease. Neuroscience 2006, 139, 1343–1354, doi:10.1016/j.neuroscience.2006.01.047.
- Abisambra, J.F.; Jinwal, U.K.; Blair, L.J.; O’Leary, J.C., 3rd.; Li, Q.; Brady, S.; Wang, L.; Guidi, C.E.; Zhang, B.; Nordhues, B.A.; et al. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J. Neurosci. 2013, 33, 9498–9507, doi:10.1523/JNEUROSCI.5397-12.2013.
- Radford, H.; Moreno, J.A.; Verity, N.; Halliday, M.; Mallucci, G.R. PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 2015, 130, 633–642, doi:10.1007/s00401-015-1487-z.
- Segev, Y.; Barrera, I.; Ounallah-Saad, H.; Wibrand, K.; Sporild, I.; Livne, A.; Rosenberg, T.; David, O.; Mints, M.; Bramham, C.R.; et al. PKR inhibition rescues memory deficit and ATF4 overexpression in ApoE epsilon4 human replacement mice. J. Neurosci. 2015, 35, 12986–12993, doi:10.1523/JNEUROSCI.5241-14.2015.
- O’Connor, T.; Sadleir, K.R.; Maus, E.; Velliquette, R.A.; Zhao, J.; Cole, S.L.; Eimer, W.A.; Hitt, B.; Bembinster, L.A.; Lammich, S.; et al. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron 2008, 60, 988–1009, doi:10.1016/j.neuron.2008.10.047.
- Yang, W.; Zhou, X.; Zimmermann, H.R.; Cavener, D.R.; Klann, E.; Ma, T. Repression of the eIF2alpha kinase PERK alleviates mGluR-LTD impairments in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2016, 41, 19–24, doi:10.1016/j.neurobiolaging.2016.02.005.
- Costa-Mattioli, M.; Gobert, D.; Stern, E.; Gamache, K.; Colina, R.; Cuello, C.; Sossin, W.; Kaufman, R.; Pelletier, J.; Rosenblum, K.; et al. eIF2alpha phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell 2007, 129, 195–206, doi:10.1016/j.cell.2007.01.050.
- Baleriola, J.; Walker, C.A.; Jean, Y.Y.; Crary, J.F.; Troy, C.M.; Nagy, P.L.; Hengst, U. Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell 2014, 158, 1159–1172, doi:10.1016/j.cell.2014.07.001.
- DuBoff, B.; Götz, J.; Feany, M.B. Tau promotes neurodegeneration via DRP1 mislocalization In Vivo. Neuron 2012, 75, 618–632, doi:10.1016/j.neuron.2012.06.026.Tau.
- Biswas, J.; Gupta, S.; Verma, D.K.; Gupta, P.; Singh, A.; Tiwari, S.; Goswami, P.; Sharma, S.; Singh, S. Involvement of glucose related energy crisis and endoplasmic reticulum stress: Insinuation of streptozotocin induced Alzheimer’s like pathology. Cell. Signal. 2018, 42, 211–226, doi:10.1016/j.cellsig.2017.10.018.
- Cui, W.; Wang, S.; Wang, Z.; Wang, Z.; Sun, C.; Zhang, Y. Inhibition of PTEN attenuates endoplasmic reticulum stress and apoptosis via activation of PI3K/AKT pathway in Alzheimer’s disease. Neurochem. Res. 2017, 42, 3052–3060, doi:10.1007/s11064-017-2338-1.
- Lin, L.; Liu, G.; Yang, L. Crocin improves cognitive behavior in rats with Alzheimer’s disease by regulating endoplasmic reticulum stress and apoptosis. Biomed. Res. Int. 2019, 2019, doi:10.1155/2019/9454913.
- Reinhardt, S.; Schuck, F.; Grosgen, S.; Riemenschneider, M.; Hartmann, T.; Postina, R.; Grimm, M.; Endres, K. Unfolded protein response signaling by transcription factor XBP-1 regulates ADAM10 and is affected in Alzheimer’s disease. FASEB J. 2014, 28, 978–997, doi:10.1096/fj.13-234864.
- Gerakis, Y.; Dunys, J.; Bauer, C.; Checler, F. Aβ42 oligomers modulate β-secretase through an XBP-1s-dependent pathway involving HRD1. Sci. Rep. 2016, 6, 37436, doi:10.1038/srep37436.
- Bussiere, R.; Oules, B.; Mary, A.; Vaillant-Beuchot, L.; Martin, C.; El Manaa, W.; Vallee, D.; Duplan, E.; Paterlini-Brechot, P.; Alves Da Costa, C.; et al. Upregulation of the sarco-endoplasmic reticulum calcium ATPase 1 truncated isoform plays a pathogenic role in Alzheimer’s disease. Cells 2019, 8, doi:10.3390/cells8121539.
- Mori, T.; Hayashi, T.; Hayashi, E.; Su, T.P. Sigma-1 receptor chaperone at the ER-Mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS ONE 2013, 8, doi:10.1371/journal.pone.0076941.
- Takeda, K.; Nagashima, S.; Shiiba, I.; Uda, A.; Tokuyama, T.; Ito, N.; Fukuda, T.; Matsushita, N.; Ishido, S.; Iwawaki, T.; et al. MITOL prevents ER stress-induced apoptosis by IRE1alpha ubiquitylation at ER-mitochondria contact sites. EMBO J. 2019, 38, e100999, doi:10.15252/embj.2018100999.
- Son, S.M.; Byun, J.; Roh, S.E.; Kim, S.J.; Mook-Jung, I. Reduced IRE1alpha mediates apoptotic cell death by disrupting calcium homeostasis via the InsP3 receptor. Cell Death Dis. 2014, 5, e1188, doi:10.1038/cddis.2014.129.
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666.
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355.
- Lee, J.H.; Won, S.M.; Suh, J.; Son, S.J.; Moon, G.J.; Park, U.J.; Gwag, B.J. Induction of the unfolded protein response and cell death pathway in Alzheimer’s disease, but not in aged Tg2576 mice. Exp. Mol. Med. 2010, 42, 386–394, doi:10.3858/emm.2010.42.5.040.
- Cissé, M.; Duplan, E.; Lorivel, T.; Dunys, J.; Bauer, C.; Meckler, X.; Gerakis, Y.; Lauritzen, I.; Checler, F. The transcription factor XBP1s restores hippocampal synaptic plasticity and memory by control of the Kalirin-7 pathway in Alzheimer model. Mol. Psychiatry 2017, 22, 1562–1575, doi:10.1038/mp.2016.152.
- Casas-Tinto, S.; Zhang, Y.; Sanchez-Garcia, J.; Gomez-Velazquez, M.; Rincon-Limas, D.E.; Fernandez-Funez, P. The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum. Mol. Genet. 2011, 20, 2144–2160, doi:10.1093/hmg/ddr100.
- Waldherr, S.M.; Strovas, T.J.; Vadset, T.A.; Liachko, N.F.; Kraemer, B.C. Constitutive XBP-1s-mediated activation of the endoplasmic reticulum unfolded protein response protects against pathological tau. Nat. Commun. 2019, 10, 4443, doi:10.1038/s41467-019-12070-3.
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 2007, 27, 53–66, doi:10.1016/j.molcel.2007.06.011.
- Chami, M.; Gozuacik, D.; Saigo, K.; Capiod, T.; Falson, P.; Lecoeur, H.; Urashima, T.; Beckmann, J.; Gougeon, M.L.; Claret, M.; et al. Hepatitis B virus-related insertional mutagenesis implicates SERCA1 gene in the control of apoptosis. Oncogene 2000, 19, 2877–2886.
- Chami, M.; Gozuacik, D.; Lagorce, D.; Brini, M.; Falson, P.; Peaucellier, G.; Pinton, P.; Lecoeur, H.; Gougeon, M.L.; le Maire, M.; et al. SERCA1 truncated proteins unable to pump calcium reduce the endoplasmic reticulum calcium concentration and induce apoptosis. J. Cell Biol. 2001, 153, 1301–1314.
- Chami, M.; Oules, B.; Szabadkai, G.; Tacine, R.; Rizzuto, R.; Paterlini-Brechot, P. Role of SERCA1 truncated isoform in the proapoptotic calcium transfer from ER to mitochondria during ER stress. Mol. Cell 2008, 32, 641–651, doi:10.1016/j.molcel.2008.11.014.


