 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Cristina Solé | + 5818 word(s) | 5818 | 2020-12-16 10:29:06 | | | |
| 2 | Rita Xu | -2450 word(s) | 3368 | 2021-01-08 04:30:56 | | |
Video Upload Options
MicroRNAs (miRNAs) are endogenous small non-coding RNA molecules that regulate the gene expression at a post-transcriptional level and participate in maintaining the correct cell homeostasis and functioning. Different specific profiles have been identified in lesional skin from autoimmune cutaneous diseases, and their deregulation cause aberrant control of biological pathways, contributing to pathogenic conditions.
1. Introduction
MicroRNAs, also known as miRs or miRNAs, are small, highly conserved, non-coding RNA sequences that range from 19 to 25 nucleotides [1]. In recent years, thousands of miRNAs have been discovered employing new advances in molecular biology and bioinformatics, achieving relevance in translational research. miRNA biogenesis has been broadly investigated to establish that most miRNAs are transcribed from DNA sequences in the nucleus by RNA polymerase II (Pol II). Drosha, a member of the RNase III family, with protein DiGeorge syndrome critical region gene 8 (DGCR8), constitute the microprocessor complex that cleaves the primary miRNAs to generate a 70-nucleotide sequence called miRNA precursor [2][3]. This is exported by exportin-5 to the cytoplasm and then processed by RNase III endonuclease dicer. After processing, the terminal loop is removed resulting in a miRNA duplex that will be incorporated into the argonaute (AGO) family of proteins. The directionality of the miRNA determines the name of the mature form. Both 5-p and 3-p strands can be loaded into the AGO proteins; however, the selection of the 5p or 3p is based on the thermodynamic stability at 5’ ends of the miRNA duplex or a 5’ U at nucleotide position 1. Usually, strands with lower 5’ stability or 5’ uracil are preferentially loaded into AGO and are named “guide strands”. The unloaded strand is called a “passenger strand”, and it is degraded. After the miRNA duplex is unwound, it is incorporated into the RNA-induced silencing complex (RISC), forming the minimal miRNA-induced silencing complex (miRISC), and then, the miRNA 20 nucleotide’s (nt’s) mature form is able to recognise and target complementary mRNA sequences (Figure 1).
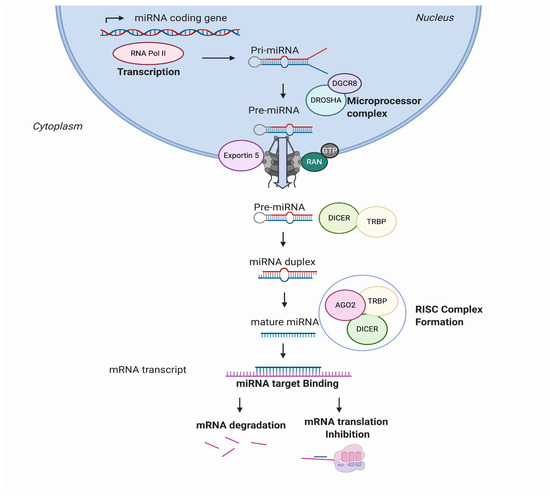
Figure 1. MicroRNA (miRNA) biogenesis and regulation of gene expression. miRNAs are transcribed from the genome into a pre-miRNA. The pre-miRNA is a smaller stem-looped structure that is transported from the nucleus to the cytoplasm by Exportin 5. Once in the cytoplasm, it is cleaved by DICER and TRBP and results into a small mRNA duplex that is around 20–25 nucleotides of length. The duplex is separated, and one of the strands is incorporated into the RISC, formed by AGO member proteins. The mature miRNA is then generated and binds specifically the mRNA transcript by complementary target recognition. The mRNA–miRNA union prevents the mRNA translation or leads into mRNA degradation and subsequent gene silencing. AGO: argonaute protein family and RISC: RNA-induced silencing complex.
MicroRNAs can modulate the gene expression at the same cell where they are being synthetised, or they can be secreted, enveloped in extracellular vesicles (EVs), transported from a parental cell to neighbouring cells and regulate important biological functions in the recipient cells [4]. Moreover, a single miRNA may have multiple target genes, and a single gene may be targeted by multiple miRNAs [5], making them a powerful system for modulating and adjusting the gene expression, as they approximately regulate around 60% of all the protein-coding genes [6].
miRNAs are involved in development, organogenesis, proliferation and apoptosis, among other cell processes [7][8]. Under normal physiological conditions, microRNAs are regulating correct cell functions. However, in disease, microRNAs may change, inducing an altered gene expression that leads into an aberrant phenotype [9].
2. Role of miRNAs in the Skin Pathogenesis of Cutaneous Immune Disorders
Skin is the largest organ in the human body, and its development and morphogenesis require a highly regulated and undisrupted miRNA profile. miRNAs’ role in skin physiology is well-known [10][11], as they are involved in epidermal and dermal proliferation, pigmentation, aging, wound healing, skin microbiome and skin immunity, among other processes [12]. Recent findings show that miRNAs have a role in skin carcinogenesis [13] and in the pathogenesis of chronic inflammatory skin diseases, presenting lesional specific miRNA expression profiles that differ from healthy skin [14][15][16]. A better understanding of the role of miRNAs in autoimmune cutaneous diseases will enhance our knowledge of skin disease pathology. In this section, the most important miRNAs associated with psoriasis, cutaneous lupus disease (CLE) and atopic dermatitis (AD) are described with special emphasis on their role in the disease pathogenesis.
2.1. Psoriasis
Psoriasis is the most prevalent chronic inflammatory skin disease, with an estimated prevalence in adults ranging from 0.91% to 8.5%, varying by country and ethnicity [17]. Genetic and environmental factors in connection with abnormal regulation of the immune system are thought to be involved in pathogenesis of the disease. It is characterised by hyperproliferation and altered differentiation of epidermal keratinocytes and leukocyte infiltration—predominantly, neutrophils, myeloid cells and T cells, causing the secretion of inflammatory mediators such as TNF-α, interferon-γ (IFN-γ), interleukin (IL)-1, IL-17 and IL-22, which contribute to psoriatic inflammation [18]. It has been identified that the IL-23/IL-17 axis is the primary signalling pathway, leading to characteristic molecular and cellular changes in psoriatic skin [18]. It is widely accepted that psoriasis is a consequence of an impaired crosstalk between the immune system and the structural cells of the skin. Several studies have been conducted to reveal the role of miRNAs in psoriasis (Table 1 and Figure 2), highlighting the value of miRNA analysis. The role of miR-203, miR-31, miR-146a, miR-155-5p and miR-21 are described below.
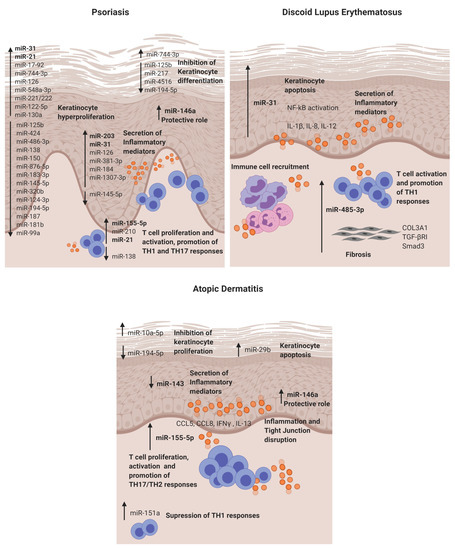
Figure 2. Dysregulated miRNAs involved in Psoriasis, discoid lupus erythematosus and atopic dermatitis and their roles in the disease pathogenesis. DLE: discoid lupus erythematosus and AD: atopic dermatitis.
Table 1. Differentially expressed mRNAs in skin immune diseases. Tissue/cell/fluids in which microRNAs (miRNAs) are found dysregulated, miRNA expression, validated experimentally target genes, and their biological role in the skin are detailed.
|
miRNA |
Condition |
Tissue/Cell/Fluid |
Expression |
Target Genes |
Biological Role |
Ref. |
|
miR-203 |
Psoriasis |
Keratinocytes |
Upregulated |
SOCS3 NR1H3 PPARG |
Keratinocyte proliferation, modulation of cytokines: TNF-α, IL-24 and IL-8 and angiogenesis. |
|
|
miR-31 |
Psoriasis DLE |
Keratinocytes Blood DMSCs
|
Upregulated (Blood and Keratinocytes) Downregulated (DMSCs) |
PPP6C STK40 |
Keratinocyte proliferation and apoptosis. Promotes Inflammation via NFKB1 activation and chemokine and cytokine production (CXCL1, CXCL5, IL-8, IL-1B and IL-12). Neutrophile and intermediate monocyte recruitment. |
|
|
miR-146a |
Psoriasis AD |
Keracinocytes Serum |
Upregulated |
CCL5 IRAK1 CARD10 |
Protective role disminishing keratinocyte proliferation and inflammation supressing IL-17, CCL5, CCL8 and IFNγ. |
|
|
miR-155 |
Psoriasis AD |
Keratinocytes Blood T cells |
Upregulated |
CTLA4 PKIA GATA3 CASP3 |
Promoted epidermal proliferation, inflammation, TJ disruption and inhibits apoptosis. T cell proliferation and promotion of TH17 responses. |
|
|
miR-21 |
Psoriasis |
Keratinocytes Blood T cells |
Upregulated |
CASP8 SMAD7 |
T cell activation and inhibition of apoptosis. Keratinocyte proliferation and inflammation (IL-1β, CCL5 and CXCL10). |
|
|
miR-125b |
Psoriasis |
Keratinocytes |
Downregulated |
FGFR2 |
Keratinocyte proliferation and differentiation. |
[45] |
|
miR-424 |
Psoriasis |
Keratinocytes Serum |
Downregulated |
n.d. |
Keratinocyte proliferation via MEK1/cyclin E1. |
[46] |
|
miR-486-3p |
Psoriasis |
Keratinocytes |
Downegulated |
K17 |
Keratinocyte proliferation. |
[47] |
|
miR-138 |
Psoriasis |
Keratinocytes |
Downregulated |
K17 |
Keratinocyte proliferation and apoptosis reduction. |
[48] |
|
miR-744-3p |
Psoriasis |
Keratinocytes |
Upregulated |
KLLN |
Keratinocyte proliferation and differentiation. |
[49] |
|
miR-150 |
Psoriasis |
Keratinocytes |
Downregulated |
HIF1A VEGFA |
Keratinocyte proliferation in hypoxic conditions. |
[50] |
|
miR-876-5p |
Psoriasis |
Skin Blood |
Downregulated |
ANG-1 |
HaCAT proliferation via PI3K/AKT, cell adhesion and invasion. |
[51] |
|
miR-183-3p |
Psoriasis |
Keratinocytes |
Downregulated |
GAB1 |
Proliferation and migration of HaCat cells. |
[52] |
|
miR-548a-3p |
Psoriasis |
Keratinocytes |
Upregulated |
PPP3R1 |
Keratinocyte proliferation. |
[53] |
|
miR-217 |
Psoriasis |
Keratinocytes |
Downregulated |
GFHL2 |
Keratinocyte differentiation. |
[54] |
|
miR-4516 |
Psoriasis |
Keratinocytes |
Downregulation |
FN1 ITGA9 |
Accelerated migration, resistance to apoptosis and differentiation. |
[55] |
|
miR-194-5p |
Psoriasis AD |
Keratinocytes |
Downregulated |
GRHL2 HS3ST2 |
Keratinocyte proliferation and inhibition of differentiation. |
[56] |
|
miR-187 |
Psoriasis |
Keratinocytes |
Downregulated |
CD276 |
Keratinocute proliferation. |
[57] |
|
miR-99a |
Psoriasis |
Keratinocytes |
Downregulated |
FZD5/FDZ8 |
Keratinocyte proliferation. |
[58] |
|
miR-130a |
Psoriasis |
Keratinocytes |
Upregulated |
STK40 |
Apoptosis inhibition and cell viability and migration promotion. Direct regulation NFKB pathway via STK40 and inditect regulation of JNK/MAPK pathway via SOX9. |
[59] |
|
miR-122-5p |
Psoriasis |
Keratinocytes |
Upregulated |
SPRY2 |
Keratinocyte proliferation. |
[60] |
|
miR-126 |
Psoriasis |
Keratinocytes |
Upregulated |
n.d. |
Keraintocyte proliferation and inflammation increasing TNFa, IFNg, IL17A, IL-22 and decreasing IL-10. Apoptosis inhibition. |
[61] |
|
miR-145-5p |
Psoriasis |
Keratinocytes |
Downregulated |
MLK3 |
Cell proliferation and chemokine secretion via NF-kB and STAT 3 activation. |
[62] |
|
miR-17-92 |
Psoriasis |
Keratinocytes |
Upregulated |
CDKN2B |
Keratinocyte proliferation and immune chemotaxis via secretion CXCL9, CXCL10, supression of SOCS1 and STAT1 activation. |
[63] |
|
miR-320b |
Psoriasis |
Keratinocytes |
Downregulation |
AKT3 |
Keratinocyte proliferation and modulation of STAT3 and SAPK/JNKsingaling pathways. |
[64] |
|
miR-124-3p |
Psoriasis |
Keratinocytes |
Downregulated |
FGFR2 |
Keratinocyte prolfieration, migration and inflammation. |
[65] |
|
miR-184 |
Psoriasis |
Keratinocytes |
Upregulated |
AGO2 |
Cytokine dependent depletion of AGO2. |
[66] |
|
miR-221/222 |
Psoriasis |
Keratinocytes |
Upregulated |
n.d. |
Keratinocyte and immune cells proliferation. |
[67] |
|
miR-181-b |
Psoriasis |
Keratinocytes |
Downregulated |
TLR4 |
Inflammation and keratinocyte proliferation. |
[68] |
|
miR-1307-3p |
Psoriasis |
Keratinocytes |
Upregulated |
n.d. |
Induces inflammatory mediators IL-8, IL-6 and CCL20. |
[69] |
|
miR-381-3p |
Psoriasis |
Keratinocytes (EVs) |
Upregulated |
FOXO1 UBR5 |
Crosslink with T cells inducing TH1/TH17 polarisation. |
[70] |
|
miR-210 |
Psoriasis |
CD4+ T cells |
Upregulated |
FOXP3 |
Induces immune T cell dysfunction. |
[71] |
|
miR-138 |
Psoriasis |
CD4+ T cells |
Downregulated |
RUNX3 |
Modulation of TH1/TH2 balance. |
[72] |
|
miR-485-3p |
DLE |
T cells Fibroblasts |
Upregulated |
PPARGC1A |
T cell activation and promotion of fibrotic processes. |
[16] |
|
miR-10a-5p |
AD |
Keratinocytes |
Upregulated |
HAS3 |
Inhibitis keratinocyte proliferation. |
[73] |
|
miR-29b |
AD |
keratinocytes |
Upregulated |
BCL2 |
Keratinocyte apoptosis. |
[74] |
|
miR-223 |
AD |
Blood |
Upregulated |
n.d. |
Upregulation of HNMT indirectly to degrade excessive histamine. |
[75] |
|
miR-151a |
AD |
Blood |
Upregulated |
IL12RB2 |
Regulation of TH1 cytokines (IL-2, IFN γ). |
[76] |
|
miR-143 |
AD |
Keratinocytes |
Downregulatd |
IL13RA1 |
Regulation of IL-13 activitu and TH2 inflammation. |
[77] |
The first study in 2007 that reported a distinctive skin miRNA signature in psoriasis was published by E Sonkoly et al. [14]. The study identified miR-203 as a keratinocyte-derived microRNA related to inflammation by targeting the SOCS3 gene. After that, further studies have confirmed the direct targeting [19] and its role in the regulation of psoriatic cytokines such as TNF-α, IL-24 and IL-8 in keratinocytes [20][21]. Moreover, in vitro experiments showed that miR-203 expression is upregulated after IL-17 stimulation in HaCat cells and that miR-203 is involved in the activation of the JAK2/STAT3 signalling pathway, which contributes to VEGF secretion and the perpetuation of pathological angiogenesis [19]. Recently, it has been described that miR-203 negatively regulates keratinocyte proliferation through the direct targeting of NR1H3 and PPARG [22]. Therefore, in psoriasis, the data suggest that miR-203 may be involved in skin epidermal hyperplasia, inflammation and angiogenesis (Figure 2).
MiR-31 is known to be involved in normal skin physiology by regulating keratinocyte growth and hair differentiation [78]. High miR-31 levels can be detected in blood and lesional psoriatic epidermis, and its pathogenic role is primarily based on NF-κB signalling alteration [23][24]. NF-κB is a crucial mediator in the pathogenesis of psoriasis and participates in inflammation, cell proliferation, differentiation and apoptosis. Serine/threonine kinase 40 (STK40), a negative regulator of NF-κB signalling, has been identified as a direct target for miR-31 [25]. The study demonstrated that miR-31 promotes NF-κB via STK40 targeting and leads to the secretion of CXLC1, CXCL8, CXCL5 and IL-1β, which promote vascular endothelial cell activation and attract leukocytes via chemotaxis into the skin. Primary keratinocytes treated with TGFβ1, which is highly expressed in psoriatic skin, showed an upregulation of miR-31 [25]. This effect was also observed when keratinocytes were treated with psoriatic-relevant cytokines: IL-6, IL-22, interferon-γ (IFN-γ) and TNF-α [24], demonstrating its importance in epidermal inflammation. This miRNA is also involved in keratinocyte proliferation, as in vivo studies showed that miR-31 promotes epidermal hyperplasia via the direct targeting of PPP6C, a negative regulator of the G1-S phase progression in the cell cycle [24]. Endothelin-1, a peptide involved in cell proliferation and leukocyte chemotaxis, has been positively associated with high levels of miR-31 in blood [23]. MiR-31 may play a role in dermal mesenchymal stem cells (DMSCs) [79], as low levels in DMSCs of psoriasis patients versus healthy controls are found, but this needs further investigation. Taken together, miR-31 has a crucial role in psoriasis by promoting epidermal proliferation and inflammation in lesion sites.
MiR-146a is overexpressed in lesional skin and peripheral blood mononuclear cells (PBMCs) from psoriasis patients [14][26]. It is known for its negative role in epidermal inflammation by targeting NF-κB mediators IRAK1 and CARD10 and chemotactic attractant CCL5 [27][28][29]. Xia et al. [26] showed that high levels of miR-146a in the skin and in PBMCs of psoriasis patients positively correlate with IL-17 levels in the skin and serum, respectively. However, target gene IRAK1 was downregulated in PBMCs but not in the skin, showing the asynchronous expression of target genes in local lesions and peripheral PBMCs. In vivo studies using mice models of Psoriasis showed that miR-146a inhibition promoted earlier psoriasis-like onset, epidermal hyperproliferation, IL-17 skin inflammation and IL-8 secretion with the increased infiltration of neutrophils at lesion sites.
MiR-155-5p has been shown upregulated in blood and psoriatic lesional skin [30][31]. It is involved in the keratinocyte cell cycle, as in vitro studies showed that miR-155 inhibition decreases keratinocyte proliferation and increases the expression of PTEN, PIP3, AKT, BAX and BCL2 apoptotic genes [32]. Another study supported this finding by showing that miR-155-5p overexpression impairs keratinocyte apoptosis possibly by targeting CASP3, a validated direct target of miRNA-155-5p [33]. This miRNA is also involved in inflammation, as keratinocytes treated with TNF-α upregulated its expression. Moreover, when cells were stimulated with LPS and overexpressed miR-155-5p, there was an increase of TLR4; NF-κB proteins together with the levels of secreted TNF-α and IL-18, IL-6 and IL-1β via inflammasome NLRP3/CASP1 activation [34]. CXCL8 is also upregulated in miR-155-5p-overexpressed keratinocytes via the GATA3/IL37 axis [35]. Elevated miR-155 levels have also been observed in DMSCs [36]; however, further research is needed to establish its role. Overall, miR-155-5p is involved in keratinocyte proliferation, apoptosis and inflammation in psoriasis.
Finally, epidermal cells and infiltrated T cells in psoriasis lesions have shown increased miR-21 expression [41]. In vitro, it is regulated by LncRNA MEG3 and regulates keratinocyte proliferation via the direct targeting of CASP8 [42]. It also promotes proliferation by regulating the AKT/PI3K and TGFβ signalling pathways [43][44]. Regarding its role in inflammation, UVB-exposure promoted miR-21-3p upregulation in keratinocytes. This upregulation led to the production of proinflammatory cytokines IL-6 and IL-1β and chemokines CCL5 and CXCL10 in keratinocytes [44]. The expression of miR-21 is increased in both TH1 and TH2 differentiated T cells after activation with anti-CD3 and anti-CD28, indicating that this miR is involved in T-cell activation regardless the T-cell subtype. Moreover, it has an antiapoptotic effect on the activated T cells [41].
Therefore, this miRNA can contribute to psoriasis pathogenesis by modulating the cell cycle and inflammation in keratinocytes and T cells.
Twenty-seven further miRNAs have also been described in psoriasis pathogenesis. They are detailed in Table 1 [45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62][63][64][65][66][67][68][69][70][71][72].
2.2. Cutaneous Lupus Erythematosus (CLE)
Cutaneous lupus erythematosus (CLE) is an autoimmune chronic disease that includes a broad range of dermatologic manifestations. CLE is divided into several subtypes, but discoid lupus erythematosus (DLE) is consistently reported as the most common subtype, and this may be because, as a chronic disorder, it is easier to identify compared to the more evanescent and nonscarring acute cutaneous and subacute cutaneous forms (SCLE) [80]. The CLE overall prevalence is estimated to be around 73.24 per 100,000 according to several USA studies [81]. The pathogenesis of CLE is not completely understood. It seems to be multifactorial and involves genetic predisposition, environmental triggers and abnormalities in the immune response. Findings indicate that UVB may act as a trigger, promoting skin damage and keratinocyte apoptosis. There may be a defective apoptosis/cell clearance, and the immune system is activated against autoantigens.
CLE lesions share extensive lymphocytic infiltrates with a high predominance of CD4 T cells with an imbalance towards T-helper 1, cytotoxic CD8+ T cells, as well as interferon type 1 signature and proinflammatory cytokines, IL-1α, IL-1β, IL-8, TNF-α and IL-6 [82]. To date, we have published the only microRNA study in CLE—in particular, discoid lupus [16]. The study identified in DLE lesions a different microRNA signature (miR-31 and miR-485-3p) when compared to nonlesional sites. The relevant identified miRNAs and their potential role in CLE pathogenesis are detailed below.
MiR-31 was identified as a keratinocyte-derived miR located in the DLE lesional epidermis. It is involved in epidermal apoptosis by promoting the upregulation of apoptotic genes (BIM, BAX, P53 and CASP3) when overexpressed. Moreover, as in previous reports, we also found that it enhances NF-κB activation and the secretion of inflammatory cytokines such as IL-1β, IL-12 and IL-8 in keratinocytes. The crosslink between keratinocytes and lymphocytes is of critical importance in cutaneous autoimmune diseases, and it was found that miR-31 promotes the attraction of neutrophils and intermediate monocytes; therefore, it enhances the recruitment of immune cells into the DLE lesion sites, perpetuating inflammation.
MiR-485-3p was found in the infiltrating lymphocytes and fibroblasts in DLE lesions. It is involved in the activation of CD4+ and CD8+ T cells and, also, in promoting fibrosis by enhancing fibrotic genes SMAD3, COL3A1 and TGFβR in fibroblasts. This fibrosis may occur, as miR-485-3p may be targeting peroxisome PPARGC1A, which is known for exerting a protective function of fibrosis development [83] and was found downregulated in fibroblasts that overexpressed miR-485-3p. Studies showing the direct target of PPARGC1A by miR-485-3p support this finding [71].
2.3. Atopic Dermatitis (AD)
Atopic dermatitis is a complex, systemic inflammatory disorder associated with a variety of clinical features [72]. It is the most common chronic inflammatory skin disease, with a prevalence of 15–20% of children and 1–3% of adults worldwide. It has high heritability; occurs frequently with other atopic diseases (asthma, allergic rhinitis and food allergies) and its incidence has increased two to three-fold in recent years in industrialised countries [73].
AD is characterised by an epidermal barrier disruption, activation of a T-helper 2 response and alteration of the skin microbiome [72]. IgE and eosinophils are increased, which, in turn, are thought to boost inflammation and skin damage through the production of reactive oxygen species, inflammatory cytokines and the release of toxic granule proteins [74]. miRNA expression profiles in the skin lesions of AD patients have been determined by microarray. The elevated expression of let-7i, miR-24, miR-27a, miR-29a, miR-193a, miR-199a and miR-222 was reported [15]. Gu et al. also reported a multitude of dysregulated miRNAs (e.g., upregulation: miR-4270, miR-211, miR-4529-3p and miR-29b and downregulation: miR-184, miR-135a and miR-4454) in AD skin biopsies [76]. From the identified miRNAs, we describe below the functional role of some of the most relevant in the skin pathogenesis of AD.
MiR-155-5p in AD lesional skin is predominantly expressed in infiltrating immune cells. This miR plays a role in the regulation of allergen-induced inflammation by targeting CTLA4, a negative regulator of T-cell activation [15]. It affects T-cell proliferation and differentiation by shifting towards a TH17 response [84]. The expression of this miR has been analysed in different disease stages in an AD mice model, and it was found to be increased in the elicited phase of the disease compared to controls [85]. Increased levels of miR-155-5p have also been detected in vitro in HaCAT cells stimulated with TNF-α, and it promotes inflammation and epithelial tight junction (TJ) changes by the direct binding of PKIA [86]. Taken together, miR-155-5p promotes T-cell activation, epidermal inflammation and TJ disruption in AD.
Previous studies have demonstrated that miR 146a is involved in the inflammatory response of atopic dermatitis (AD). MiR-146a expression is increased in keratinocytes and the chronic lesional skin of patients with AD expression. MiR-146a may have an anti-inflammatory role, alleviating chronic skin inflammation in atopic dermatitis through the suppression of innate immune responses in keratinocytes. It inhibited the expression of numerous proinflammatory factors, including IFN-γ-inducible and AD-associated genes CCL5, CCL8, and ubiquitin D (UBD) in human primary keratinocytes stimulated with IFN-γ, TNF-α or IL-1β. Studies demonstrated that miR-146a-mediated suppression in allergic skin inflammation partially occurs through the direct targeting of the upstream NF-κB signal transducers caspase recruitment domain, containing protein 10 and IL-1 receptor-associated kinase 1. In addition, CCL5 was identified as a novel, direct target of miR-146a . It is worth mentioning that the upregulation of miR-10a-5p, miR-29b, miR-223 and miR-151a have also been described in the inflammatory response and keratinocyte apoptosis for AD patients [73][74][75][76] (Table 1).
Finally, miR-143 has been found downregulated in the lesional skin from AD patients [77]. It targets IL-13 receptor alpha 1 (IL13R), modulating IL-13 activity. IL-13 is a cytokine involved in TH2 responses that is highly expressed in AD skin lesions. Therefore, miR-143 may contribute to AD pathogenesis by favouring TH2 responses.
References
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355.
- Baek, D.; Villén, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of microRNAs on protein output. Nature 2008, 455, 64–71.
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297.
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density. Cell Biol. 2011, 13, 423–433.
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human MicroRNA targets. PLoS Biol. 2004, 2, e363.
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105.
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Cell-cycle control of microRNA-mediated translation regulation. Cell Cycle 2008, 7, 1545–1549.
- Leung, A.K.L.; Sharp, P.A. Function and localization of microRNAs in mammalian cells. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2006; Volume 71, pp. 29–38.
- Soifer, H.S.; Rossi, J.J.; Saetrom, P. MicroRNAs in disease and potential therapeutic applications. Ther. 2007, 15, 2070–2079.
- Andl, T.; Murchison, E.P.; Liu, F.; Zhang, Y.; Yunta-Gonzalez, M.; Tobias, J.W.; Andl, C.D.; Seykora, J.T.; Hannon, G.J.; Millar, S.E. The miRNA-processing enzyme dicer is essential for the morphogenesis and maintenance of hair follicles. Biol. 2006, 16, 1041–1049.
- Yi, R.; O’Carroll, D.; Pasolli, H.A.; Zhang, Z.; Dietrich, F.S.; Tarakhovsky, A.; Fuchs, E. Morphogenesis in skin is governed by discrete sets of differentially expressed miRNAs. Genet. 2006, 38, 356–362.
- Yi, R.; Fuchs, E. MicroRNA-mediated control in the skin. Cell Death Differ. 2010, 17, 229–235.
- Neagu, M.; Constantin, C.; Cretoiu, S.M.; Zurac, S. miRNAs in the Diagnosis and Prognosis of Skin Cancer. Cell Dev. Biol. 2020, 8, 71.
- Sonkoly, E.; Wei, T.; Janson, P.C.; Sääf, A.; Lundeberg, L.; Tengvall-Linder, M.; Norstedt, G.; Alenius, H.; Homey, B.; Scheynius, A.; et al. MicroRNAs: Novel regulators involved in the pathogenesis of psoriasis? PLoS ONE 2007, 2, e610.
- Sonkoly, E.; Janson, P.; Majuri, M.L.; Savinko, T.; Fyhrquist, N.; Eidsmo, L.; Xu, N.; Meisgen, F.; Wei, T.; Bradley, M.; et al. MiR-155 is overexpressed in patients with atopic dermatitis and modulates T-cell proliferative responses by targeting cytotoxic T lymphocyte-associated antigen 4. Allergy Clin. Immunol. 2010, 126, 581–920.
- Solé, C.; Domingo, S.; Ferrer, B.; Moliné, T.; Ordi-Ros, J.; Cortés-Hernández, J. MicroRNA Expression Profiling Identifies miR-31 and miR-485-3p as Regulators in the Pathogenesis of Discoid Cutaneous Lupus. Investig. Dermatol. 2019, 139, 51–61.
- Parisi, R.; Symmons, D.P.; Griffiths, C.E.; Ashcroft, D.M. Identification and Management of Psoriasis and Associated ComorbidiTy (IMPACT) project team. Global epidemiology of psoriasis: A systematic review of incidence and prevalence. Investig. Dermatol. 2013, 133, 377–385.
- Lowes, M.A.; Suárez-Fariñas, M.; Krueger, J.G. Immunology of psoriasis. Rev. Immunol. 2014, 32, 227–255.
- Xu, Y.; Ji, Y.; Lan, X.; Gao, X.; Chen, H.D.; Geng, L. miR‑203 contributes to IL‑17‑induced VEGF secretion by targeting SOCS3 in keratinocytes. Med. Rep. 2017, 16, 8989–8996.
- Primo, M.N.; Bak, R.O.; Schibler, B.; Mikkelsen, J.G. Regulation of pro-inflammatory cytokines TNFα and IL24 by microRNA-203 in primary keratinocytes. Cytokine 2012, 60, 741–748.
- Wei, T.; Xu, N.; Meisgen, F.; Ståhle, M.; Sonkoly, E.; Pivarcsi, A. Interleukin-8 is regulated by miR-203 at the posttranscriptional level in primary human keratinocytes. J. Dermatol. 2013, 19, 1997.
- Xiao, Y.; Wang, H.; Wang, C.; Zeng, B.; Tang, X.; Zhang, Y.; Peng, Y.; Luo, M.; Huang, P.; Yang, Z. miR-203 promotes HaCaT cell overproliferation through targeting LXR-α and PPAR-γ. Cell Cycle 2020, 19, 1928–1940.
- Borska, L.; Andrys, C.; Chmelarova, M.; Kovarikova, H.; Krejsek, J.; Hamakova, K.; Beranek, M.; Palicka, V.; Kremlacek, J.; Borsky, P.; et al. Roles of miR-31 and endothelin-1 in psoriasis vulgaris: Pathophysiological functions and potential biomarkers. Res. 2017, 66, 987–992.
- Yan, S.; Xu, Z.; Lou, F.; Zhang, L.; Ke, F.; Bai, J.; Liu, Z.; Liu, J.; Wang, H.; Zhu, H.; et al. NF-κB-induced microRNA-31 promotes epidermal hyperplasia by repressing protein phosphatase 6 in Nat. Commun. 2015, 6, 7652.
- Xu, N.; Meisgen, F.; Butler, L.M.; Han, G.; Wang, X.J.; Söderberg-Nauclér, C.; Ståhle, M.; Pivarcsi, A.; Sonkoly, E. MicroRNA-31 is overexpressed in psoriasis and modulates inflammatory cytokine and chemokine production in keratinocytes via targeting serine/threonine kinase 40. Immunol. 2013, 190, 678–688.
- Xia, P.; Fang, X.; Zhang, Z.H.; Huang, Q.; Yan, K.X.; Kang, K.F.; Han, L.; Zheng, Z.Z. Dysregulation of miRNA146a versus IRAK1 induces IL-17 persistence in the psoriatic skin lesions. Lett. 2012, 148, 151–162.
- Crone, S.G.; Jacobsen, A.; Federspiel, B.; Bardram, L.; Krogh, A.; Lund, A.H.; Friis-Hansen, L. microRNA-146a inhibits G protein-coupled receptor-mediated activation of NF-kappaB by targeting CARD10 and COPS8 in gastric cancer. Cancer 2012, 11, 71.
- Hung, P.S.; Liu, C.J.; Chou, C.S.; Kao, S.Y.; Yang, C.C.; Chang, K.W.; Chiu, T.H.; Lin, S.C. miR-146a enhances the oncogenicity of oral carcinoma by concomitant targeting of the IRAK1, TRAF6 and NUMB genes. PLoS ONE 2013, 8, e79926.
- Rebane, A.; Runnel, T.; Aab, A.; Maslovskaja, J.; Rückert, B.; Zimmermann, M.; Plaas, M.; Kärner, J.; Treis, A.; Pihlap, M.; et al. MicroRNA-146a alleviates chronic skin inflammation in atopic dermatitis through suppression of innate immune responses in keratinocytes. Allergy Clin. Immunol. 2014, 134, 836–847.
- García-Rodríguez, S.; Arias-Santiago, S.; Blasco-Morente, G.; Orgaz-Molina, J.; Rosal-Vela, A.; Navarro, P.; Magro-Checa, C.; Martínez-López, A.; Ruiz, J.C.; Raya, E.; et al. Increased expression of microRNA-155 in peripheral blood mononuclear cells from psoriasis patients is related to disease activity. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 312–322.
- El-Komy, M.; Amin, I.; El-Hawary, M.S.; Saadi, D.; Shaker, O. Upregulation of the miRNA-155, miRNA-210, and miRNA-20b in psoriasis patients and their relation to IL-17. J. Immunopathol. Pharmacol. 2020, 34, 2058738420933742.
- Xu, L.; Leng, H.; Shi, X.; Ji, J.; Fu, J.; Leng, H. MiR-155 promotes cell proliferation and inhibits apoptosis by PTEN signaling pathway in the psoriasis. Biomed. Pharmacother. 2017, 90, 524–530.
- Soonthornchai, W.; Tangtanatakul, P.; Meephansan, J.; Ruchusatsawat, K.; Reantragoon, R.; Hirankarn, N.; Wongpiyabovorn, J. Down-regulation of miR-155 after treatment with narrow-band UVB and methotrexate associates with apoptosis of keratinocytes in psoriasis. Asian Pac. J. Allergy Immunol. 2019, doi:10.12932/AP-031218-0451.
- Luo, Q.; Zeng, J.; Li, W.; Lin, L.; Zhou, X.; Tian, X.; Liu, W.; Zhang, L.; Zhang, X. Silencing of miR‑155 suppresses inflammatory responses in psoriasis through inflammasome NLRP3 regulation. J. Mol. Med. 2018, 42, 1086–1095.
- Wang, H.; Zhang, Y.; Luomei, J.; Huang, P.; Zhou, R.; Peng, Y. The miR-155/GATA3/IL37 axis modulates the production of proinflammatory cytokines upon TNF-α stimulation to affect psoriasis development. Dermatol. 2020, doi:10.1111/exd.14117.
- Hou, R.X.; Liu, R.F.; Zhao, X.C.; Jia, Y.R.; An, P.; Hao, Z.P.; Li, J.Q.; Li, X.H.; Yin, G.H.; Zhang, K.M. Increased miR-155-5p expression in dermal mesenchymal stem cells of psoriatic patients: Comparing the microRNA expression profile by microarray. Genet Mol. Res. 2016, 15, doi:10.4238/gmr.15038631.
- Lou, C.; Xiao, M.; Cheng, S.; Lu, X.; Jia, S.; Ren, Y.; Li, Z. MiR-485-3p and miR-485-5p suppress breast cancer cell metastasis by inhibiting PGC-1α expression. Cell Death Dis. 2016, 7, e2159.
- Weidinger, S.; Beck, L.A.; Bieber, T.; Kabashima, K.; Irvine, A.D. Atopic dermatitis. Rev. Dis. Primers 2018, 4, 1.
- Nutten, S. Atopic dermatitis: Global epidemiology and risk factors. Nutr. Metab. 2015, 66, 8–16.
- Liu, F.T.; Goodarzi, H.; Chen, H.Y. IgE, mast cells, and eosinophils in atopic dermatitis. Rev. Allergy Immunol. 2011, 41, 298–310.
- Meisgen, F.; Xu, N.; Wei, T.; Janson, P.C.; Obad, S.; Broom, O.; Nagy, N.; Kauppinen, S.; Kemény, L.; Ståhle, M.; et al. MiR-21 is up-regulated in psoriasis and suppresses T cell apoptosis. Dermatol. 2012, 21, 312–314.
- Jia, H.Y.; Zhang, K.; Lu, W.J.; Xu, G.W.; Zhang, J.F.; Tang, Z.L. LncRNA MEG3 influences the proliferation and apoptosis of psoriasis epidermal cells by targeting miR-21/caspase-8. BMC Mol. Cell. Biol. 2019, 20, 46.
- Yang, C.; Luo, L.; Bai, X.; Shen, K.; Liu, K.; Wang, J.; Hu, D. Highly-expressed micoRNA-21 in adipose derived stem cell exosomes can enhance the migration and proliferation of the HaCaT cells by increasing the MMP-9 expression through the PI3K/AKT pathway. Biochem. Biophys. 2020, 681, 108259.
- Degueurce, G.; D'Errico, I.; Pich, C.; Ibberson, M.; Schütz, F.; Montagner, A.; Sgandurra, M.; Mury, L.; Jafari, P.; Boda, A.; et al. Identification of a novel PPARβ/δ/miR-21-3p axis in UV-induced skin inflammation. EMBO Mol. Med. 2016, 8, 919–936.
- Xu, N.; Brodin, P.; Wei, T.; Meisgen, F.; Eidsmo, L.; Nagy, N.; Kemeny, L.; Stahle, M.; Sonkoly, E.; Pivarcsi, A. MiR-125b, a microRNA downregulated in psoriasis, modulates keratinocyte proliferation by targeting FGFR2. Investig. Dermatol. 2011, 131, 1521–1529.
- Ichihara, A.; Jinnin, M.; Yamane, K.; Fujisawa, A.; Sakai, K.; Masuguchi, S.; Fukushima, S.; Maruo, K.; Ihn, H. microRNA-mediated keratinocyte hyperproliferatio in psoriasis vulgaris. J. Dermatol. 2011, 165, 1003–1010.
- Jiang, M.; Sun, Z.; Dang, E.; Li, B.; Fang, H.; Li, J.; Gao, L.; Zhang, K.; Wang, G. TGFβ/SMAD/microRNA-486-3p Signaling Axis Mediates Keratin 17 Expression and Keratinocyte Hyperproliferation in Psoriasis. J. Dermatol. 2017, 137, 2177–2186.
- Feng, S.J.; Chu, R.Q.; Ma, J.; Wang, Z.X.; Zhang, G.J.; Yang, X.F.; Song, Z.; Ma, Y.Y. MicroRNA138 regulates keratin 17 protein expression to affect HaCaT cell proliferation and apoptosis by targeting hTERT in psoriasis vulgaris. Pharmacother. 2017, 85, 169–176.
- Wang, C.; Zong, J.; Li, Y.; Wang, X.; Du, W.; Li, L. MiR-744-3p regulates keratinocyte proliferation and differentiation via targeting KLLN in psoriasis. Dermatol. 2019, 28, 283–291.
- Li, Y.; Su, J.; Li, F.; Chen, X.; Zhang, G. MiR-150 regulates human keratinocyte proliferation in hypoxic conditions through targeting HIF-1α and VEGFA: Implications for psoriasis treatment. PLoS ONE 2017, 12, e0175459.
- A, R.; Yu, P.; Hao, S.; Li, Y. MiR-876-5p suppresses cell proliferation by targeting Angiopoietin-1 in the psoriasis. Pharmacother. 2018, 103, 1163–1169.
- Liu, T.; Zhang, X.; Wang, Y. miR-183-3p suppresses proliferation and migration of keratinocyte in psoriasis by inhibiting GAB1. Hereditas 2020, 157, 28.
- Zhao, X.; Li, R.; Qiao, M.; Yan, J.; Sun, Q. MiR-548a-3p Promotes Keratinocyte Proliferation Targeting PPP3R1 after Being Induced by IL-22. Inflammation 2018, 41, 496–504.
- Zhu, H.; Hou, L.; Liu, J.; Li, Z. MiR-217 is down-regulated in psoriasis and promotes keratinocyte differentiation via targeting GRHL2. Biophys. Res. Commun. 2016, 471, 169–176.
- Chowdhari, S.; Sardana, K.; Saini, N. miR-4516, a microRNA downregulated in psoriasis inhibits keratinocyte motility by targeting fibronectin/integrin α9 signaling. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3142–3152.
- Yu, X.; An, J.; Hua, Y.; Li, Z.; Yan, N.; Fan, W.; Su, C. MicroRNA-194 regulates keratinocyte proliferation and differentiation by targeting Grainyhead-like 2 in Pathol. Res. Pract. 2017, 213, 89–97.
- Tang, L.; He, S.; Zhu, Y.; Feng, B.; Su, Z.; Liu, B.; Xu, F.; Wang, X.; Liu, H.; Chutian, L.; et al. Downregulated miR-187 contributes to the keratinocytes hyperproliferation in psoriasis. Cell. Physiol. 2019, 234, 3661–3674.
- Shen, H.; Tian, Y.; Yao, X.; Liu, W.; Zhang, Y.; Yang, Z. MiR-99a inhibits keratinocyte proliferation by targeting Frizzled-5 (FZD5) / FZD8 through β-catenin signaling in psoriasis. Pharmazie 2017, 72, 461–467.
- Xiong, Y.; Chen, H.; Liu, L.; Lu, L.; Wang, Z.; Tian, F.; Zhao, Y. microRNA-130a Promotes Human Keratinocyte Viability and Migration and Inhibits Apoptosis Through Direct Regulation of STK40-Mediated NF-κB Pathway and Indirect Regulation of SOX9-Meditated JNK/MAPK Pathway: A Potential Role in Psoriasis. DNA Cell Biol. 2017, 36, 219–226.
- Jiang, M.; Ma, W.; Gao, Y.; Jia, K.; Zhang, Y.; Liu, H.; Sun, Q. IL-22-induced miR-122-5p promotes keratinocyte proliferation by targeting Sprouty2. Dermatol. 2017, 26, 368–374.
- Feng, S.; Wang, L.; Liu, W.; Zhong, Y.; Xu, S. MiR-126 correlates with increased disease severity and promotes keratinocytes proliferation and inflammation while suppresses cells’ apoptosis in psoriasis. Clin. Lab Anal. 2018, 32, e22588.
- Yan, J.J.; Qiao, M.; Li, R.H.; Zhao, X.T.; Wang, X.Y.; Sun, Q. Downregulation of miR-145-5p contributes to hyperproliferation of keratinocytes and skin inflammation in psoriasis. J. Dermatol. 2019, 180, 365–372.
- Zhang, W.; Yi, X.; An, Y.; Guo, S.; Li, S.; Song, P.; Chang, Y.; Zhang, S.; Gao, T.; Wang, G.; et al. MicroRNA-17-92 cluster promotes the proliferation and the chemokine production of keratinocytes: Implication for the pathogenesis of psoriasis. Cell Death Dis. 2018, 9, 567.
- Wang, Y.; Yu, X.; Wang, L.; Ma, W.; Sun, Q. miR-320b Is Down-Regulated in Psoriasis and Modulates Keratinocyte Proliferation by Targeting AKT3. Inflammation 2018, 41, 2160–2170.
- Xiao, Y.; Wang, C.; Zeng, B.; Tang, X.; Zhang, Y.; Xiang, L.; Mi, L.; Pan, Y.; Wang, H.; Yang, Z. miR124-3p/FGFR2 axis inhibits human keratinocyte proliferation and migration and improve the inflammatory microenvironment in psoriasis. Immunol. 2020, 122, 89–98.
- Roberts, J.C.; Warren, R.B.; Griffiths, C.E.; Ross, K. Expression of microRNA-184 in keratinocytes represses argonaute 2. Cell. Physiol. 2013, 228, 2314–2323.
- Zibert, J.R.; Løvendorf, M.B.; Litman, T.; Olsen, J.; Kaczkowski, B.; Skov, L. MicroRNAs and potential target interactions in psoriasis. Dermatol. Sci. 2010, 58, 177–185.
- Feng, C.; Bai, M.; Yu, N.Z.; Wang, X.J.; Liu, Z. MicroRNA-181b negatively regulates the proliferation of human epidermal keratinocytes in psoriasis through targeting TLR4. Cell. Mol. Med. 2017, 21, 278–285.
- Srivastava, A.; Pasquali, L.; Pivarcsi, A.; Sonkoly, E. MiR-1307 is upregulated in psoriasis keratinocytes and promotes keratinocyte inflammatory response. Presented at the 49th Annual ESDR Meeting, Bordeaux, France, (18th September, 2019).
- Jiang, M.; Fang, H.; Dang, E.; Zhang, J.; Qiao, P.; Yu, C.; Yang, A.; Wang, G. Small extracellular vesicles containing miR-381-3p from keratinocytes promotes Th1/Th17 polarization in psoriasis. Invest. Dermatol. 2020, S0022-202X, 31938–31932.
- Zhao, M.; Wang, L.T.; Liang, G.P.; Zhan, P.; Deng, X.J.; Tang, Q.; Zhai, H.; Chang, C.C.; Su, Y.W.; Lu, Q.J. Up-regulation of microRNA-210 induces immune dysfunction via targeting FOXP3 in CD4(+) T cells of psoriasis vulgaris. Immunol. 2014, 150, 22–30.
- Fu, D.; Yu, W.; Li, M.; Wang, H.; Liu, D.; Song, X.; Li, Z.; Tian, Z. MicroRNA-138 regulates the balance of Th1/Th2 via targeting RUNX3 in psoriasis. Lett. 2015, 166, 55–62.
- Vaher, H.; Runnel, T.; Urgard, E.; Aab, A.; Carreras Badosa, G.; Maslovskaja, J.; Abram, K.; Raam, L.; Kaldvee, B.; Annilo, T.; et al. miR-10a-5p is increased in atopic dermatitis and has capacity to inhibit keratinocyte proliferation. Allergy 2019, 74, 2146–2156.
- Gu, C.; Li, Y.; Wu, J.; Xu, J. IFN‐γ‐induced microRNA‐29b up‐regulation contributes to keratinocyte apoptosis in atopic dermatitis through inhibiting Bcl2L2. J. Clin. Exp. Pathol. 2017, 10, 10117–10126.
- Jia, H.Z.; Liu, S.L.; Zou, Y.F.; Chen, X.F.; Yu, L.; Wan, J.; Zhang, H.Y.; Chen, Q.; Xiong, Y.; Yu, B.; et al. MicroRNA-223 is involved in the pathogenesis of atopic dermatitis by affecting histamine-N-methyltransferase. Mol. Biol. 2018, 64, 103–107.
- Chen, X.F.; Zhang, L.J.; Zhang, J.; Dou, X.; Shao, Y.; Jia, X.J.; Zhang, W.; Yu, B. MiR-151a is involved in the pathogenesis of atopic dermatitis by regulating interleukin-12 receptor β2. Dermatol. 2018, 27, 427–432.
- Jia, Q.N.; Zeng, Y.P. Rapamycin blocks the IL-13-induced deficiency of Epidermal Barrier Related Proteins via upregulation of miR-143 in HaCaT Keratinocytes. J. Med. Sci. 2020, 17, 2087–2094.
- Luan, L.; Shi, J.; Yu, Z.; Andl, T. The major miR-31 target genes STK40 and LATS2 and their implications in the regulation of keratinocyte growth and hair differentiation. Dermatol. 2017, 26, 497–504.
- Wang, Q.; Chang, W.; Yang, X.; Cheng, Y.; Zhao, X.; Zhou, L.; Li, J.; Li, J.; Zhang, K. Levels of miR-31 and its target genes in dermal mesenchymal cells of patients with psoriasis. J. Dermatol. 2019, 58, 198–204.
- Kuhn, A.; Landmann, A. The classification and diagnosis of cutaneous lupus erythematosus. J. Autoimmun. 2014, 48, 14–19.
- Durosaro, O.; Davis, M.; Reed, K.B.; Rohlinger, A.L. Incidence of cutaneous lupus erythematosus, 1965-2005: A population-based study. Arch. Dermatol. 2009, 145, 249–253.
- Achtman, J.C.; Werth, V.P. Pathophysiology of cutaneous lupus erythematosus. Arthritis Res. Ther. 2015, 17, 182.
- Dinulovic, I.; Furrer, R.; Di Fulvio, S.; Ferry, A.; Beer, M.; Handschin, C. PGC-1α modulates necrosis, inflammatory response, and fibrotic tissue formation in injured skeletal muscle. Skelet. Muscle 2016, 6, 38.
- Ma, L.; Xue, H.B.; Wang, F.; Shu, C.M.; Zhang, J.H. MicroRNA-155 may be involved in the pathogenesis of atopic dermatitis by modulating the differentiation and function of T helper type 17 (Th17) cells. Clin. Exp. Immunol. 2015, 181, 142–149.
- Vennegaard, M.T.; Bonefeld, C.M.; Hagedorn, P.H.; Bangsgaard, N.; Løvendorf, M.B.; Ødum, N.; Woetmann, A.; Geisler, C.; Skov, L. Allergic contact dermatitis induces upregulation of identical microRNAs in humans and mice. Contact Dermat. 2012, 67, 298–305.
- Wang, X.; Chen, Y.; Yuan, W.; Yao, L.; Wang, S.; Jia, Z.; Wu, P.; Li, L.; Wei, P.; Wang, X.; et al. MicroRNA-155-5p is a key regulator of allergic inflammation, modulating the epithelial barrier by targeting PKIα. Cell Death Dis. 2019, 10, 884.


